Epithelial–mesenchymal transition
The epithelial–mesenchymal transition (EMT) is a process by which epithelial cells lose their cell polarity and cell–cell adhesion, and gain migratory and invasive properties to become mesenchymal stem cells; these are multipotent stromal cells that can differentiate into a variety of cell types. EMT is essential for numerous developmental processes including mesoderm formation and neural tube formation. EMT has also been shown to occur in wound healing, in organ fibrosis and in the initiation of metastasis in cancer progression.
| Epithelial-mesenchymal transition | |
|---|---|
Diagram showing the epithelial-mesenchymal transition | |
| Details | |
| Precursor | endoderm |
| Identifiers | |
| MeSH | D058750 |
| Anatomical terminology | |
Introduction
Epithelial–mesenchymal transition was first recognized as a feature of embryogenesis by Betty Hay in the 1980s.[1][2] EMT, and its reverse process, MET (mesenchymal-epithelial transition) are critical for development of many tissues and organs in the developing embryo, and numerous embryonic events such as gastrulation, neural crest formation, heart valve formation, secondary palate development, and myogenesis.[3] Epithelial and mesenchymal cells differ in phenotype as well as function, though both share inherent plasticity.[2] Epithelial cells are closely connected to each other by tight junctions, gap junctions and adherens junctions, have an apico-basal polarity, polarization of the actin cytoskeleton and are bound by a basal lamina at their basal surface. Mesenchymal cells, on the other hand, lack this polarization, have a spindle-shaped morphology and interact with each other only through focal points.[4] Epithelial cells express high levels of E-cadherin, whereas mesenchymal cells express those of N-cadherin, fibronectin and vimentin. Thus, EMT entails profound morphological and phenotypic changes to a cell.[5]
Based on the biological context, EMT has been categorized into 3 types: developmental (Type I), fibrosis[6] and wound healing (Type II), and cancer (Type III).[7][8][9]
Inducers
Loss of E-cadherin is considered to be a fundamental event in EMT. Many transcription factors (TFs) that can repress E-cadherin directly or indirectly can be considered as EMT-TF (EMT inducing TFs). SNAI1/Snail 1, SNAI2/Snail 2 (also known as Slug), ZEB1, ZEB2, TCF3 and KLF8 (Kruppel-like factor 8) can bind to the E-cadherin promoter and repress its transcription, whereas factors such as Twist, Goosecoid, TCF4 (also known as E2.2), homeobox protein SIX1 and FOXC2 (fork-head box protein C2) repress E-cadherin indirectly.[10][11] SNAIL and ZEB factors bind to E-box consensus sequences on the promoter region, while KLF8 binds to promoter through GT boxes. These EMT-TFs not only directly repress E-cadherin, but also repress transcriptionally other junctional proteins, including claudins and desmosomes, thus facilitating EMT. On the other hand, transcription factors such as grainyhead-like protein 2 homologue (GRHL2), and ETS-related transcription factors ELF3 and ELF5 are downregulated during EMT and are found to actively drive MET when overexpressed in mesenchymal cells.[12][13] Since EMT in cancer progression recaptures EMT in developmental programs, many of the EMT-TFs are involved in promoting metastatic events.[14][15]
Several signaling pathways (TGF-β, FGF, EGF, HGF, Wnt/beta-catenin and Notch) and hypoxia may induce EMT.[7][16][17] In particular, Ras-MAPK has been shown to activate Snail and Slug.[18][19][20] Slug triggers the steps of desmosomal disruption, cell spreading, and partial separation at cell–cell borders, which comprise the first and necessary phase of the EMT process. On the other hand, Slug cannot trigger the second phase,[21] which includes the induction of cell motility, repression of the cytokeratin expression, and activation of vimentin expression.[22] Snail and Slug are known to regulate the expression of p63 isoforms, another transcription factor that is required for proper development of epithelial structures.[23] The altered expression of p63 isoforms reduced cell–cell adhesion and increased the migratory properties of cancer cells. The p63 factor is involved in inhibiting EMT and reduction of certain p63 isoforms may be important in the development of epithelial cancers.[24] Some of them are known to regulate the expression of cytokeratins.[25] The phosphatidylinositol 3' kinase (PI3K)/AKT axis, Hedgehog signaling pathway, nuclear factor-kappaB and Activating Transcription Factor 2 have also been implicated to be involved in EMT.[26][27][28][29]
Wnt signaling pathway regulates EMT in gastrulation, cardiac valve formation and cancer.[30] Activation of Wnt pathway in breast cancer cells induces the EMT regulator SNAIL and upregulates the mesenchymal marker, vimentin. Also, active Wnt/beta-catenin pathway correlates with poor prognosis in breast cancer patients in the clinic. Similarly, TGF-β activates the expression of SNAIL and ZEB to regulate EMT in heart development, palatogenesis, and cancer. The breast cancer bone metastasis has activated TGF-β signaling, which contributes to the formation of these lesions.[31] However, on the other hand, p53, a well-known tumor suppressor, represses EMT by activating the expression of various microRNAs – miR-200 and miR-34 that inhibit the production of protein ZEB and SNAIL, and thus maintain the epithelial phenotype.[32]
In development and wound healing
After the initial stage of embryogenesis, the implantation of the embryo and the initiation of placenta formation are associated with EMT. The trophoectoderm cells undergo EMT to facilitate the invasion of endometrium and appropriate placenta placement, thus enabling nutrient and gas exchange to the embryo. Later in embryogenesis, during gastrulation, EMT allows the cells to ingress in a specific area of the embryo – the primitive streak in amniotes, and the ventral furrow in Drosophila. The cells in this tissue express E-cadherin and apical-basal polarity.[33] Since gastrulation is a very rapid process, E-cadherin is repressed transcriptionally by Twist and SNAI1 (commonly called Snail), and at the protein level by P38 interacting protein. The primitive streak, through invagination, further generates mesoendoderm, which separates to form a mesoderm and an endoderm, again through EMT. Mesenchymal cells from the primitive streak participate also in the formation of many epithelial mesodermal organs, such as notochord as well as somites, through the reverse of EMT, i.e. mesenchymal–epithelial transition. Amphioxus forms an epithelial neural tube and dorsal notochord but does not have the EMT potential of the primitive streak. In higher chordates, the mesenchyme originates out of the primitive streak migrates anteriorly to form the somites and participate with neural crest mesenchyme in formation of the heart mesoderm.
In vertebrates, epithelium and mesenchyme are the basic tissue phenotypes. During embryonic development, migratory neural crest cells are generated by EMT involving the epithelial cells of the neuroectoderm. As a result, these cells dissociate from neural folds, gain motility, and disseminate to various parts of the embryo, where they differentiate to many other cell types. Also, craniofacial crest mesenchyme that forms the connective tissue forming the head and face, is formed by neural tube epithelium by EMT.[34] EMT takes place during the construction of the vertebral column out of the extracellular matrix, which is to be synthesized by fibroblasts and osteoblasts that encircle the neural tube. The major source of these cells are sclerotome and somite mesenchyme as well as primitive streak. Mesenchymal morphology allows the cells to travel to specific targets in the embryo, where they differentiate and/or induce differentiation of other cells.[34][35]
During wound healing, keratinocytes at the border of the wound undergo EMT and undergo re-epithelialization or MET when the wound is closed. Snail2 expression at the migratory front influences this state, as its overexpression accelerates wound healing. Similarly, in each menstrual cycle, the ovarian surface epithelium undergoes EMT during post-ovulatory wound healing.[36]
In cancer progression and metastasis
Initiation of metastasis requires invasion, which is enabled by EMT.[37][38] Carcinoma cells in a primary tumor lose cell-cell adhesion mediated by E-cadherin repression and break through the basement membrane with increased invasive properties, and enter the bloodstream through intravasation. Later, when these circulating tumor cells (CTCs) exit the bloodstream to form micro-metastases, they undergo MET for clonal outgrowth at these metastatic sites. Thus, EMT and MET form the initiation and completion of the invasion-metastasis cascade.[39] At this new metastatic site, the tumor may undergo other processes to optimize growth. For example, EMT has been associated with PD-L1 expression, particularly in lung cancer. Increased levels of PD-L1 suppresses the immune system which allows the cancer to spread more easily. [40]
EMT confers resistance to oncogene-induced premature senescence. Twist1 and Twist2, as well as ZEB1 protects human cells and mouse embryonic fibroblasts from senescence. Similarly, TGF-β can promote tumor invasion and evasion of immune surveillance at advanced stages. When TGF-β acts on activated Ras-expressing mammary epithelial cells, EMT is favored and apoptosis is inhibited.[41] This effect can be reversed by inducers of epithelial differentiation, such as GATA-3.[42]
EMT has been shown to be induced by androgen deprivation therapy in metastatic prostate cancer.[14] Activation of EMT programs via inhibition of the androgen axis provides a mechanism by which tumor cells can adapt to promote disease recurrence and progression. Brachyury, Axl, MEK, and Aurora kinase A are molecular drivers of these programs, and inhibitors are currently in clinical trials to determine therapeutic applications.[14] Oncogenic PKC-iota can promote melanoma cell invasion by activating Vimentin during EMT. PKC-iota inhibition or knockdown resulted an increase E-cadherin and RhoA levels while decreasing total Vimentin, phosphorylated Vimentin (S39) and Par6 in metastatic melanoma cells. These results suggested that PKC-ι is involved in signaling pathways which upregulate EMT in melanoma.[43][44]
EMT has been indicated to be involved in acquiring drug resistance. Gain of EMT markers was found to be associated with the resistance of ovarian carcinoma epithelial cell lines to paclitaxel. Similarly, SNAIL also confers resistance to paclitaxel, adriamycin and radiotherapy by inhibiting p53-mediated apoptosis.[45] Furthermore, inflammation, that has been associated with the progression of cancer and fibrosis, was recently shown to be related to cancer through inflammation-induced EMT.[46] Consequently, EMT enables cells to gain a migratory phenotype, as well as induce multiple immunosuppression, drug resistance, evasion of apoptosis mechanisms.
Some evidence suggests that cells that undergo EMT gain stem cell-like properties, thus giving rise to Cancer Stem Cells (CSCs). Upon transfection by activated Ras, a subpopulation of cells exhibiting the putative stem cell markers CD44high/CD24low increases with the concomitant induction of EMT.[47] Also, ZEB1 is capable of conferring stem cell-like properties, thus strengthening the relationship between EMT and stemness. Thus, EMT may present increased danger to cancer patients, as EMT not only enables the carcinoma cells to enter the bloodstream, but also endows them with properties of stemness which increases tumorigenic and proliferative potential.[48]
However, recent studies have further shifted the primary effects of EMT away from invasion and metastasis, toward resistance to chemotherapeutic agents. Research on breast cancer and pancreatic cancer both demonstrated no difference in cells' metastatic potential upon acquisition of EMT.[49][50] These are in agreement with another study showing that the EMT transcription factor TWIST actually requires intact adherens junctions in order to mediate local invasion in breast cancer.[51] The effects of EMT and its relationship to invasion and metastasis may therefore be highly context specific.
In urothelial carcinoma cell lines overexpression of HDAC5 inhibits long-term proliferation but can promote epithelial-to-mesenchymal transition (EMT).[52]
Platelets in cancer EMT
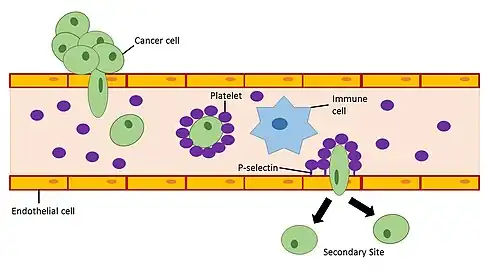
Platelets in the blood have the ability to initiate the induction of EMT in cancer cells. When platelets are recruited to a site in the blood vessel they can release a variety of growth factors (PDGF,[53] VEGF,[54] Angiopoietin-1[55]) and cytokines including the EMT inducer TGF-β.[56] The release of TGF-β by platelets in blood vessels near primary tumors enhances invasiveness and promotes metastasis of cancer cells in the tumor.[57] Studies looking at defective platelets and reduced platelet counts in mouse models have shown that impaired platelet function is associated with decreased metastatic formation.[58][59] In humans, platelet counts and thrombocytosis within the upper end of the normal range have been associated with advanced, often metastatic, stage cancer in cervical cancer,[60] ovarian cancer,[61] gastric cancer,[62] and esophageal cancer.[63] Although a great deal of research has been applied to studying interactions between tumor cells and platelets, a cancer therapy targeting this interaction has not yet been established.[64] This may be in part due to the redundancy of prothrombotic pathways which would require the use of multiple therapeutic approaches in order to prevent pro-metastatic events via EMT induction in cancer cells by activated platelets.
To improve the chances for the development of a cancer metastasis, a cancer cell must avoid detection and targeting by the immune system once it enters the bloodstream. Activated platelets have the ability to bind glycoproteins and glycolipids (P-selectin ligands such as PSGL-1) on the surface of cancer cells to form a physical barrier that protects the cancer cell from natural killer cell-mediated lysis in the bloodstream.[65] Furthermore, activated platelets promote the adhesion of cancer cells to activated endothelial cells lining blood vessels using adhesion molecules present on platelets.[66][64] P-selectin ligands on the surface of cancer cells remain to be elucidated and may serve as potential biomarkers for disease progression in cancer.[64]
Therapeutics targeting cancer EMT
Many studies have proposed that induction of EMT is the primary mechanism by which epithelial cancer cells acquire malignant phenotypes that promote metastasis.[67] Drug development targeting the activation of EMT in cancer cells has thus become an aim of pharmaceutical companies.[68]
Small molecule inhibitors
Small molecules that are able to inhibit TGF-β induced EMT are under development.[68] Silmitasertib (CX-4945) is a small molecule inhibitor of protein kinase CK2, which has been supported to be linked with TGF-β induced EMT, and is currently in clinical trials for cholangiocarcinoma (bile duct cancer), as well as in preclinical development for hematological and lymphoid malignancies.[69][70] In January 2017, Silmitasertib was granted orphan drug status by the U.S. Food and Drug Administration for cholangiocarcinoma and is currently in phase II study. Silmitasertib is being developed by Senhwa Biosciences.[71] Another small molecule inhibitor Galunisertib (LY2157299) is a potent TGF-β type I receptor kinase inhibitor that was demonstrated to reduce the size, the growth rate of tumors, and the tumor forming potential in triple negative breast cancer cell lines using mouse xenografts.[72] Galunisertib is currently being developed by Lilly Oncology and is in phase I/II clinical trials for hepatocellular carcinoma, unresectable pancreatic cancer, and malignant glioma.[73] Small molecule inhibitors of EMT are suggested to not act as a replacement for traditional chemotherapeutic agents but are likely to display the greatest efficacy in treating cancers when used in conjunction with them.
Antagomirs and microRNA mimics have gained interest as a potential source of therapeutics to target EMT induced metastasis in cancer as well as treating many other diseases.[74] Antagomirs were first developed to target miR-122, a microRNA that was abundant and specific to the liver, and this discovery has led to the development of other antagomirs that can pair with specific microRNAs present in the tumor microenvironment or in the cancer cells.[75][73] A microRNA mimic to miR-655 was found to suppress EMT through the targeting of EMT inducing transcription factor ZEB1 and TGF-β receptor 2 in a pancreatic cancer cell line. Overexpression of the miR-655 mimic in the Panc1 cancer cell line upregulated the expression of E-cadherin and suppressed the migration and invasion of mesenchymal-like cancer cells.[76] The use of microRNA mimics to suppress EMT has expanded to other cancer cell lines and holds potential for clinical drug development.[74] However, microRNA mimics and antagomirs suffer from a lack of stability in vivo and lack an accurate delivery system to target these molecules to the tumor cells or tissue for treatment.[77] Improvements to antagomir and microRNA mimic stability through chemical modifications such as locked nucleic acid (LNA) oligonucleotides or peptide nucleic acids (PNA) can prevent the fast clearing of these small molecules by RNases.[77][74] Delivery of antagomirs and microRNA mimics into cells by enclosing these molecules in liposome-nanoparticles has generated interest however liposome structures suffer from their own drawbacks that will need to be overcome for their effective use as a drug delivery mechanism.[77] These drawbacks of liposome-nanoparticles include nonspecific uptake by cells and induction of immune responses.[78] The role that microRNAs play in cancer development and metastasis is under much scientific investigation and it is yet to be demonstrated whether microRNA mimics or antagomirs may serve as standard clinical treatments to suppress EMT or oncogenic microRNAs in cancers.[74]
Generation of endocrine progenitor cells from pancreatic islets
Similar to generation of Cancer Stem Cells, EMT was demonstrated to generate endocrine progenitor cells from human pancreatic islets.[79] Initially, the human islet-derived progenitor cells (hIPCs) were proposed to be better precursors since β-cell progeny in these hIPCs inherit epigenetic marks that define an active insulin promoter region.[80] However, later, another set of experiments suggested that labelled β-cells de-differentiate to a mesenchymal-like phenotype in vitro, but fail to proliferate; thus initiating a debate in 2007.[81][82][83]
Since these studies in human islets lacked lineage-tracing analysis, these findings from irreversibly tagged beta cells in mice were extrapolated to human islets. Thus, using a dual lentiviral and genetic lineage tracing system to label β-cells, it was convincingly demonstrated that adult human islet β-cells undergo EMT and proliferate in vitro.[84][85] Also, these findings were confirmed in human fetal pancreatic insulin-producing cells, and the mesenchymal cells derived from pancreatic islets can undergo the reverse of EMT – MET – to generate islet-like cell aggregates.[86] Thus, the concept of generating progenitors from insulin-producing cells by EMT or generation of Cancer Stem Cells during EMT in cancer may have potential for replacement therapy in diabetes, and call for drugs targeting inhibition of EMT in cancer.[86]
Partial EMT or a hybrid E/M phenotype
Not all cells undergo a complete EMT, i.e. losing their cell-cell adhesion and gaining solitary migration characteristics. Instead, most cells undergo partial EMT, a state in which they retain some epithelial traits such as cell-cell adhesion or apico-basal polarity, and gain migratory traits, thus cells in this hybrid epithelial/mesenchymal (E/M) phenotype are endowed with special properties such as collective cell migration.[51][87][88][30][89][90][91][92] Two mathematical models have been proposed, attempting to explain the emergence of this hybrid E/M phenotype,[89][91] and its highly likely that different cell lines adopt different hybrid state(s), as shown by experiments in MCF10A, HMLE and H1975 cell lines.[90][93] Although a hybrid E/M state has been referred to as 'metastable' or transient, recent experiments in H1975 cells suggest that this state can be stably maintained by cells.[94]
See also
References
- Kong D, Li Y, Wang Z, Sarkar FH (February 2011). "Cancer Stem Cells and Epithelial-to-Mesenchymal Transition (EMT)-Phenotypic Cells: Are They Cousins or Twins?". Cancers. 3 (1): 716–29. doi:10.3390/cancers30100716. PMC 3106306. PMID 21643534.
- Lamouille S, Xu J, Derynck R (March 2014). "Molecular mechanisms of epithelial-mesenchymal transition". Nature Reviews. Molecular Cell Biology. 15 (3): 178–96. doi:10.1038/nrm3758. PMC 4240281. PMID 24556840.
- Thiery JP, Acloque H, Huang RY, Nieto MA (November 2009). "Epithelial-mesenchymal transitions in development and disease". Cell. 139 (5): 871–90. doi:10.1016/j.cell.2009.11.007. PMID 19945376. S2CID 10874320.
- Thiery JP, Sleeman JP (February 2006). "Complex networks orchestrate epithelial-mesenchymal transitions". Nature Reviews. Molecular Cell Biology. 7 (2): 131–42. doi:10.1038/nrm1835. PMID 16493418. S2CID 8435009.
- Francou A, Anderson KV (2020). "The Epithelial-to-Mesenchymal Transition in Development and Cancer". Annual Review of Cancer Biology. 4: 197–220. doi:10.1146/annurev-cancerbio-030518-055425. PMC 8189433. PMID 34113749.
- Phua YL, Martel N, Pennisi DJ, Little MH, Wilkinson L (April 2013). "Distinct sites of renal fibrosis in Crim1 mutant mice arise from multiple cellular origins". The Journal of Pathology. 229 (5): 685–96. doi:10.1002/path.4155. PMID 23224993. S2CID 22837861.
- Kalluri R, Weinberg RA (June 2009). "The basics of epithelial-mesenchymal transition". The Journal of Clinical Investigation. 119 (6): 1420–8. doi:10.1172/JCI39104. PMC 2689101. PMID 19487818.
- Sciacovelli M, Frezza C (October 2017). "Metabolic reprogramming and epithelial-to-mesenchymal transition in cancer". The FEBS Journal. 284 (19): 3132–3144. doi:10.1111/febs.14090. PMC 6049610. PMID 28444969.
- Li L, Li W (June 2015). "Epithelial-mesenchymal transition in human cancer: comprehensive reprogramming of metabolism, epigenetics, and differentiation". Pharmacology & Therapeutics. 150: 33–46. doi:10.1016/j.pharmthera.2015.01.004. PMID 25595324.
- Peinado H, Olmeda D, Cano A (2007). "Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype?". Nature Reviews Cancer. 7 (6): 415–428. doi:10.1038/nrc2131. hdl:10261/81769. PMID 17508028. S2CID 25162191.
- Yang J, Weinberg RA (2008). "Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis". Dev Cell. 14 (6): 818–829. doi:10.1016/j.devcel.2008.05.009. PMID 18539112.
- De Craene B, Berx G (2013). "Regulatory networks defining EMT during cancer initiation and progression". Nature Reviews Cancer. 13 (2): 97–110. doi:10.1038/nrc3447. PMID 23344542. S2CID 13619676.
- Chakrabarti R, Hwang J, Andres Blanco M, Wei Y, Lukačišin M, Romano RA, Smalley K, Liu S, Yang Q, Ibrahim T, Mercatali L, Amadori D, Haffty BG, Sinha S, Kang Y (2012). "Elf5 inhibits the epithelial-mesenchymal transition in mammary gland development and breast cancer metastasis by transcriptionally repressing Snail2". Nat Cell Biol. 14 (11): 1212–1222. doi:10.1038/ncb2607. PMC 3500637. PMID 23086238.
- Nouri M, Ratther E, Stylianou N, Nelson CC, Hollier BG, Williams ED (2014). "Androgen-targeted therapy-induced epithelial mesenchymal plasticity and neuroendocrine transdifferentiation in prostate cancer: an opportunity for intervention". Front Oncol. 4: 370. doi:10.3389/fonc.2014.00370. PMC 4274903. PMID 25566507.
- Puisieux A, Brabletz T, Caramel J (June 2014). "Oncogenic roles of EMT-inducing transcription factors". Nature Cell Biology. 16 (6): 488–94. doi:10.1038/ncb2976. PMID 24875735. S2CID 5226347.
- Zhang L, Huang G, Li X, Zhang Y, Jiang Y, Shen J, et al. (March 2013). "Hypoxia induces epithelial-mesenchymal transition via activation of SNAI1 by hypoxia-inducible factor -1α in hepatocellular carcinoma". BMC Cancer. 13: 108. doi:10.1186/1471-2407-13-108. PMC 3614870. PMID 23496980.
- "Epithelial-Mesenchymal Transition | GeneTex". www.genetex.com. Retrieved 28 October 2019.
- Horiguchi K, Shirakihara T, Nakano A, Imamura T, Miyazono K, Saitoh M (January 2009). "Role of Ras signaling in the induction of snail by transforming growth factor-beta". The Journal of Biological Chemistry. 284 (1): 245–53. doi:10.1074/jbc.m804777200. PMID 19010789.
- Ciruna B, Rossant J (July 2001). "FGF signaling regulates mesoderm cell fate specification and morphogenetic movement at the primitive streak". Developmental Cell. 1 (1): 37–49. doi:10.1016/s1534-5807(01)00017-x. PMID 11703922.
- Lu Z, Ghosh S, Wang Z, Hunter T (December 2003). "Downregulation of caveolin-1 function by EGF leads to the loss of E-cadherin, increased transcriptional activity of beta-catenin, and enhanced tumor cell invasion". Cancer Cell. 4 (6): 499–515. doi:10.1016/s1535-6108(03)00304-0. PMID 14706341.
- Savagner P, Yamada KM, Thiery JP (June 1997). "The zinc-finger protein slug causes desmosome dissociation, an initial and necessary step for growth factor-induced epithelial-mesenchymal transition". The Journal of Cell Biology. 137 (6): 1403–19. doi:10.1083/jcb.137.6.1403. PMC 2132541. PMID 9182671.
- Boyer B, Tucker GC, Vallés AM, Franke WW, Thiery JP (October 1989). "Rearrangements of desmosomal and cytoskeletal proteins during the transition from epithelial to fibroblastoid organization in cultured rat bladder carcinoma cells". The Journal of Cell Biology. 109 (4 Pt 1): 1495–509. doi:10.1083/jcb.109.4.1495. PMC 2115780. PMID 2677020.
- Herfs M, Hubert P, Suarez-Carmona M, Reschner A, Saussez S, Berx G, et al. (April 2010). "Regulation of p63 isoforms by snail and slug transcription factors in human squamous cell carcinoma". The American Journal of Pathology. 176 (4): 1941–9. doi:10.2353/ajpath.2010.090804. PMC 2843482. PMID 20150431.
- Lindsay J, McDade SS, Pickard A, McCloskey KD, McCance DJ (February 2011). "Role of DeltaNp63gamma in epithelial to mesenchymal transition". The Journal of Biological Chemistry. 286 (5): 3915–24. doi:10.1074/jbc.M110.162511. PMC 3030392. PMID 21127042.
- Boldrup L, Coates PJ, Gu X, Nylander K (December 2007). "DeltaNp63 isoforms regulate CD44 and keratins 4, 6, 14 and 19 in squamous cell carcinoma of head and neck". The Journal of Pathology. 213 (4): 384–91. doi:10.1002/path.2237. PMID 17935121. S2CID 21891189.
- Larue L, Bellacosa A (November 2005). "Epithelial-mesenchymal transition in development and cancer: role of phosphatidylinositol 3' kinase/AKT pathways". Oncogene. 24 (50): 7443–54. doi:10.1038/sj.onc.1209091. PMID 16288291.
- Vlahopoulos SA, Logotheti S, Mikas D, Giarika A, Gorgoulis V, Zoumpourlis V (April 2008). "The role of ATF-2 in oncogenesis". BioEssays. 30 (4): 314–27. doi:10.1002/bies.20734. PMID 18348191. S2CID 678541.
- Huber MA, Beug H, Wirth T (December 2004). "Epithelial-mesenchymal transition: NF-kappaB takes center stage". Cell Cycle. 3 (12): 1477–80. doi:10.4161/cc.3.12.1280. PMID 15539952.
- Katoh Y, Katoh M (September 2008). "Hedgehog signaling, epithelial-to-mesenchymal transition and miRNA (review)". International Journal of Molecular Medicine. 22 (3): 271–5. PMID 18698484.
- Micalizzi DS; Farabaugh SM; Ford HL (2010). "Epithelial-Mesenchymal Transition in Cancer: Parallels between Normal Development and Tumor Progression". J Mammary Gland Biol Neoplasia. 15 (2): 117–134. doi:10.1007/s10911-010-9178-9. PMC 2886089. PMID 20490631.
- Kang Y, He W, Tulley S, Gupta GP, Serganova I, Chen CR, Manova-Todorova K, Blasberg R, Gerald WL, Massagué J (2005). "Breast cancer bone metastasis mediated by the Smad tumor suppressor pathway". PNAS. 102 (39): 13909–14. Bibcode:2005PNAS..10213909K. doi:10.1073/pnas.0506517102. PMC 1236573. PMID 16172383.
- Chang C, Chao C, Xia W, Yang J, Xiong Y, Li C, Yu W, Rehman SK, Hsu JL, Lee H, Liu M, Chen C, Yu D, Hung M (2011). "p53 regulates epithelial-mesenchymal transition (EMT) and stem cell properties through modulating miRNAs". Nat Cell Biol. 13 (3): 317–323. doi:10.1038/ncb2173. PMC 3075845. PMID 21336307.
- Lim R, Thiery JP (2012). "Epithelial-mesenchymal transitions: insights from development". Development. 139 (19): 3471–3486. doi:10.1242/dev.071209. PMID 22949611.
- Hay ED (2005). "The mesenchymal cell, its role in the embryo, and the remarkable signaling mechanisms that create it". Dev. Dyn. 233 (3): 706–20. doi:10.1002/dvdy.20345. PMID 15937929. S2CID 22368548.
- Kerosuo L, Bronner-Fraser M (2012). "What is bad in cancer is good in the embryo: Importance of EMT in neural crest development". Seminars in Cell and Developmental Biology. 23 (3): 320–332. doi:10.1016/j.semcdb.2012.03.010. PMC 3345076. PMID 22430756.
- Ahmed N, Maines-Bandiera S, Quinn MA, Unger WG, Dedhar S, Auersperg N (2006). "Molecular pathways regulating EGF-induced epithelio- mesenchymal transition in human ovarian surface epithelium". Am J Physiol Cell Physiol. 290 (6): C1532–C1542. doi:10.1152/ajpcell.00478.2005. PMID 16394028. S2CID 35196500.
- Hanahan D, Weinberg RA (January 2000). "The hallmarks of cancer". Cell. 100 (1): 57–70. doi:10.1016/s0092-8674(00)81683-9. PMID 10647931. S2CID 1478778.
- Hanahan D, Weinberg RA (March 2011). "Hallmarks of cancer: the next generation". Cell. 144 (5): 646–74. doi:10.1016/j.cell.2011.02.013. PMID 21376230.
- Chaffer CL, Weinberg RA (March 2011). "A perspective on cancer cell metastasis". Science. 331 (6024): 1559–64. Bibcode:2011Sci...331.1559C. doi:10.1126/science.1203543. PMID 21436443. S2CID 10550070.
- Ye X, Weinberg RA (November 2015). "Epithelial-Mesenchymal Plasticity: A Central Regulator of Cancer Progression". Trends in Cell Biology. 25 (11): 675–686. doi:10.1016/j.tcb.2015.07.012. PMC 4628843. PMID 26437589.
- Massague J (2008). "TGFβ in cancer". Cell. 134 (2): 215–229. doi:10.1016/j.cell.2008.07.001. PMC 3512574. PMID 18662538.
- Chu IM, Lai WC, Aprelikova O, El Touny LH, Kouros-Mehr H, Green JE (2013). "Expression of GATA3 in MDA-MB-231 triple-negative breast cancer cells induces a growth inhibitory response to TGFß". PLOS ONE. 8 (4): e61125. Bibcode:2013PLoSO...861125C. doi:10.1371/journal.pone.0061125. PMC 3620110. PMID 23577196.
- Ratnayake WS, Apostolatos AH, Ostrov DA, Acevedo-Duncan M (2017). "Two novel atypical PKC inhibitors; ACPD and DNDA effectively mitigate cell proliferation and epithelial to mesenchymal transition of metastatic melanoma while inducing apoptosis". Int. J. Oncol. 51 (5): 1370–1382. doi:10.3892/ijo.2017.4131. PMC 5642393. PMID 29048609.
- Ratnayake WS, Apostolatos CA, Apostolatos AH, Schutte RJ, Huynh MA, Ostrov DA, Acevedo-Duncan M (2018). "Oncogenic PKC-ι activates Vimentin during epithelial-mesenchymal transition in melanoma; a study based on PKC-ι and PKC-ζ specific inhibitors". Cell Adhes. Migr. 12 (5): 447–463. doi:10.1080/19336918.2018.1471323. PMC 6363030. PMID 29781749.
- Kajiyama H, Shibata K, Terauchi M, Yamashita M, Ino K, Nawa A, Kikkawa F (August 2007). "Chemoresistance to paclitaxel induces epithelial-mesenchymal transition and enhances metastatic potential for epithelial ovarian carcinoma cells". International Journal of Oncology. 31 (2): 277–83. doi:10.3892/ijo.31.2.277. PMID 17611683.
- Ricciardi M, Zanotto M, Malpeli G, Bassi G, Perbellini O, Chilosi M, et al. (March 2015). "Epithelial-to-mesenchymal transition (EMT) induced by inflammatory priming elicits mesenchymal stromal cell-like immune-modulatory properties in cancer cells". British Journal of Cancer. 112 (6): 1067–75. doi:10.1038/bjc.2015.29. PMC 4366889. PMID 25668006.
- Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, Yang J, Weinberg RA (2008). "The epithelial-mesenchymal transition generates cells with properties of stem cells". Cell. 133 (4): 704–15. doi:10.1016/j.cell.2008.03.027. PMC 2728032. PMID 18485877.
- Singh A, Settleman J (2010). "EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer". Oncogene. 29 (34): 4741–4751. doi:10.1038/onc.2010.215. PMC 3176718. PMID 20531305.
- Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong ST, et al. (November 2015). "Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance". Nature. 527 (7579): 472–6. Bibcode:2015Natur.527..472F. doi:10.1038/nature15748. PMC 4662610. PMID 26560033.
- Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, et al. (November 2015). "Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer". Nature. 527 (7579): 525–530. Bibcode:2015Natur.527..525Z. doi:10.1038/nature16064. PMC 4849281. PMID 26560028.
- Shamir ER, Pappalardo E, Jorgens DM, Coutinho K, Tsai WT, Aziz K, et al. (March 2014). "Twist1-induced dissemination preserves epithelial identity and requires E-cadherin". The Journal of Cell Biology. 204 (5): 839–56. doi:10.1083/jcb.201306088. PMC 3941052. PMID 24590176.
- Jaguva Vasudevan AA, Hoffmann MJ, Beck ML, Poschmann G, Petzsch P, Wiek C, et al. (April 2019). "HDAC5 Expression in Urothelial Carcinoma Cell Lines Inhibits Long-Term Proliferation but Can Promote Epithelial-to-Mesenchymal Transition". International Journal of Molecular Sciences. 20 (9): 2135. doi:10.3390/ijms20092135. PMC 6539474. PMID 31052182.
- Kepner N, Lipton A (February 1981). "A mitogenic factor for transformed fibroblasts from human platelets". Cancer Research. 41 (2): 430–2. PMID 6256066.
- Möhle R, Green D, Moore MA, Nachman RL, Rafii S (January 1997). "Constitutive production and thrombin-induced release of vascular endothelial growth factor by human megakaryocytes and platelets". Proceedings of the National Academy of Sciences of the United States of America. 94 (2): 663–8. Bibcode:1997PNAS...94..663M. doi:10.1073/pnas.94.2.663. PMC 19570. PMID 9012841.
- Li JJ, Huang YQ, Basch R, Karpatkin S (February 2001). "Thrombin induces the release of angiopoietin-1 from platelets". Thrombosis and Haemostasis. 85 (2): 204–6. doi:10.1055/s-0037-1615677. PMID 11246533. S2CID 33522255.
- Assoian RK, Komoriya A, Meyers CA, Miller DM, Sporn MB (June 1983). "Transforming growth factor-beta in human platelets. Identification of a major storage site, purification, and characterization". The Journal of Biological Chemistry. 258 (11): 7155–60. doi:10.1016/S0021-9258(18)32345-7. PMID 6602130.
- Oft M, Heider KH, Beug H (November 1998). "TGFbeta signaling is necessary for carcinoma cell invasiveness and metastasis". Current Biology. 8 (23): 1243–52. doi:10.1016/s0960-9822(07)00533-7. PMID 9822576. S2CID 18536979.
- Bakewell SJ, Nestor P, Prasad S, Tomasson MH, Dowland N, Mehrotra M, et al. (November 2003). "Platelet and osteoclast beta3 integrins are critical for bone metastasis". Proceedings of the National Academy of Sciences of the United States of America. 100 (24): 14205–10. Bibcode:2003PNAS..10014205B. doi:10.1073/pnas.2234372100. PMC 283570. PMID 14612570.
- Camerer E, Qazi AA, Duong DN, Cornelissen I, Advincula R, Coughlin SR (July 2004). "Platelets, protease-activated receptors, and fibrinogen in hematogenous metastasis". Blood. 104 (2): 397–401. doi:10.1182/blood-2004-02-0434. PMID 15031212.
- Hernandez E, Lavine M, Dunton CJ, Gracely E, Parker J (June 1992). "Poor prognosis associated with thrombocytosis in patients with cervical cancer". Cancer. 69 (12): 2975–7. doi:10.1002/1097-0142(19920615)69:12<2975::aid-cncr2820691218>3.0.co;2-a. PMID 1591690.
- Zeimet AG, Marth C, Müller-Holzner E, Daxenbichler G, Dapunt O (February 1994). "Significance of thrombocytosis in patients with epithelial ovarian cancer". American Journal of Obstetrics and Gynecology. 170 (2): 549–54. doi:10.1016/s0002-9378(94)70225-x. PMID 8116711.
- Ikeda M, Furukawa H, Imamura H, Shimizu J, Ishida H, Masutani S, et al. (April 2002). "Poor prognosis associated with thrombocytosis in patients with gastric cancer". Annals of Surgical Oncology. 9 (3): 287–91. doi:10.1245/aso.2002.9.3.287. PMID 11923136.
- Shimada H, Oohira G, Okazumi S, Matsubara H, Nabeya Y, Hayashi H, et al. (May 2004). "Thrombocytosis associated with poor prognosis in patients with esophageal carcinoma". Journal of the American College of Surgeons. 198 (5): 737–41. doi:10.1016/j.jamcollsurg.2004.01.022. PMID 15110807.
- Erpenbeck L, Schön MP (April 2010). "Deadly allies: the fatal interplay between platelets and metastasizing cancer cells". Blood. 115 (17): 3427–36. doi:10.1182/blood-2009-10-247296. PMC 2867258. PMID 20194899.
- Palumbo JS, Talmage KE, Massari JV, La Jeunesse CM, Flick MJ, Kombrinck KW, et al. (January 2005). "Platelets and fibrin(ogen) increase metastatic potential by impeding natural killer cell-mediated elimination of tumor cells". Blood. 105 (1): 178–85. doi:10.1182/blood-2004-06-2272. PMID 15367435. S2CID 279285.
- Gay LJ, Felding-Habermann B (February 2011). "Contribution of platelets to tumour metastasis". Nature Reviews. Cancer. 11 (2): 123–34. doi:10.1038/nrc3004. PMC 6894505. PMID 21258396.
- Thiery JP (June 2002). "Epithelial-mesenchymal transitions in tumour progression". Nature Reviews. Cancer. 2 (6): 442–54. doi:10.1038/nrc822. PMID 12189386. S2CID 5236443.
- Yingling JM, Blanchard KL, Sawyer JS (December 2004). "Development of TGF-beta signalling inhibitors for cancer therapy". Nature Reviews. Drug Discovery. 3 (12): 1011–22. doi:10.1038/nrd1580. PMID 15573100. S2CID 42237691.
- Zou J, Luo H, Zeng Q, Dong Z, Wu D, Liu L (June 2011). "Protein kinase CK2α is overexpressed in colorectal cancer and modulates cell proliferation and invasion via regulating EMT-related genes". Journal of Translational Medicine. 9: 97. doi:10.1186/1479-5876-9-97. PMC 3132712. PMID 21702981.
- Gowda C, Sachdev M, Muthusami S, Kapadia M, Petrovic-Dovat L, Hartman M, et al. (2017). "Casein Kinase II (CK2) as a Therapeutic Target for Hematological Malignancies". Current Pharmaceutical Design. 23 (1): 95–107. doi:10.2174/1381612822666161006154311. PMID 27719640.
- "CX-4945 Granted Orphan Drug Designation". Oncology Times. 39 (5): 23. 10 March 2017. doi:10.1097/01.cot.0000514203.35081.69. ISSN 0276-2234.
- Bhola NE, Balko JM, Dugger TC, Kuba MG, Sánchez V, Sanders M, et al. (March 2013). "TGF-β inhibition enhances chemotherapy action against triple-negative breast cancer". The Journal of Clinical Investigation. 123 (3): 1348–58. doi:10.1172/JCI65416. PMC 3582135. PMID 23391723.
- Kothari AN, Mi Z, Zapf M, Kuo PC (15 October 2014). "Novel clinical therapeutics targeting the epithelial to mesenchymal transition". Clinical and Translational Medicine. 3: 35. doi:10.1186/s40169-014-0035-0. PMC 4198571. PMID 25343018.
- Rupaimoole R, Slack FJ (March 2017). "MicroRNA therapeutics: towards a new era for the management of cancer and other diseases". Nature Reviews. Drug Discovery. 16 (3): 203–222. doi:10.1038/nrd.2016.246. PMID 28209991. S2CID 22956490.
- Krützfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, Stoffel M (December 2005). "Silencing of microRNAs in vivo with 'antagomirs'". Nature. 438 (7068): 685–9. Bibcode:2005Natur.438..685K. doi:10.1038/nature04303. PMID 16258535. S2CID 4414240.
- Harazono Y, Muramatsu T, Endo H, Uzawa N, Kawano T, Harada K, et al. (14 May 2013). "miR-655 Is an EMT-suppressive microRNA targeting ZEB1 and TGFBR2". PLOS ONE. 8 (5): e62757. Bibcode:2013PLoSO...862757H. doi:10.1371/journal.pone.0062757. PMC 3653886. PMID 23690952.
- Rothschild SI (4 March 2014). "microRNA therapies in cancer". Molecular and Cellular Therapies. 2: 7. doi:10.1186/2052-8426-2-7. PMC 4452061. PMID 26056576.
- Lv H, Zhang S, Wang B, Cui S, Yan J (August 2006). "Toxicity of cationic lipids and cationic polymers in gene delivery". Journal of Controlled Release. 114 (1): 100–9. doi:10.1016/j.jconrel.2006.04.014. PMID 16831482.
- Gershengorn MC, Hardikar AA, Wei C, et al. (2004). "Epithelial-to-mesenchymal transition generates proliferative human islet precursor cells". Science. 306 (5705): 2261–2264. Bibcode:2004Sci...306.2261G. doi:10.1126/science.1101968. PMID 15564314. S2CID 22304970.
- Gershengorn MC, Geras-Raaka E, Hardikar AA, et al. (2005). "Are better islet cell precursors generated by epithelial-to-mesenchymal transition?". Cell Cycle. 4 (3): 380–382. doi:10.4161/cc.4.3.1538. PMID 15711124.
- Atouf F, Park CH, Pechhold K, et al. (2007). "No evidence for mouse pancreatic beta-cell epithelial-mesenchymal transition in vitro". Diabetes. 56 (3): 699–702. doi:10.2337/db06-1446. PMID 17327438.
- Chase LG, Ulloa-Montoya F, Kidder BL, et al. (2007). "Islet-derived fibroblast-like cells are not derived via epithelial-mesenchymal transition from Pdx-1 or insulin-positive cells". Diabetes. 56 (1): 3–7. doi:10.2337/db06-1165. PMID 17110468.
- Morton RA, Geras-Raaka E, Wilson LM, et al. (2007). "Endocrine precursor cells from mouse islets are not generated by epithelial-to-mesenchymal transition of mature beta cells". Mol Cell Endocrinol. 270 (1–2): 87–93. doi:10.1016/j.mce.2007.02.005. PMC 1987709. PMID 17363142.
- Russ HA, Bar Y, Ravassard P, et al. (2008). "In vitro proliferation of cells derived from adult human beta-cells revealed by cell-lineage tracing". Diabetes. 57 (6): 1575–1583. doi:10.2337/db07-1283. PMID 18316362.
- Russ HA, Ravassard P, Kerr-Conte J, et al. (2009). "Epithelial-mesenchymal transition in cells expanded in vitro from lineage-traced adult human pancreatic beta cells". PLOS ONE. 4 (7): e6417. Bibcode:2009PLoSO...4.6417R. doi:10.1371/journal.pone.0006417. PMC 2712769. PMID 19641613.
- Joglekar MV, Joglekar VM, Joglekar SV, et al. (2009). "Human fetal pancreatic insulin-producing cells proliferate in vitro". J Endocrinol. 201 (1): 27–36. doi:10.1677/joe-08-0497. PMID 19171567.
- Jolly MK, Boareto M, Huang B, Jia D, Lu M, Ben-Jacob E, et al. (1 January 2015). "Implications of the Hybrid Epithelial/Mesenchymal Phenotype in Metastasis". Frontiers in Oncology. 5: 155. arXiv:1505.07494. Bibcode:2015arXiv150507494J. doi:10.3389/fonc.2015.00155. PMC 4507461. PMID 26258068.
- Nakaya Y, Sheng G (November 2013). "EMT in developmental morphogenesis". Cancer Letters. 341 (1): 9–15. doi:10.1016/j.canlet.2013.02.037. PMID 23462225.
- Tian XJ, Zhang H, Xing J (August 2013). "Coupled reversible and irreversible bistable switches underlying TGFβ-induced epithelial to mesenchymal transition". Biophysical Journal. 105 (4): 1079–89. arXiv:1307.4732. Bibcode:2013BpJ...105.1079T. doi:10.1016/j.bpj.2013.07.011. PMC 3752104. PMID 23972859.
- Zhang J, Tian XJ, Zhang H, Teng Y, Li R, Bai F, et al. (September 2014). "TGF-β-induced epithelial-to-mesenchymal transition proceeds through stepwise activation of multiple feedback loops". Science Signaling. 7 (345): ra91. doi:10.1126/scisignal.2005304. PMID 25270257. S2CID 19143040.
- Lu M, Jolly MK, Levine H, Onuchic JN, Ben-Jacob E (November 2013). "MicroRNA-based regulation of epithelial-hybrid-mesenchymal fate determination". Proceedings of the National Academy of Sciences of the United States of America. 110 (45): 18144–9. Bibcode:2013PNAS..11018144L. doi:10.1073/pnas.1318192110. PMC 3831488. PMID 24154725.
- Savagner P (October 2010). "The epithelial-mesenchymal transition (EMT) phenomenon". Annals of Oncology. 21 (Suppl 7): vii89-92. doi:10.1093/annonc/mdq292. PMC 3379967. PMID 20943648.
- Jia D, Jolly MK, Tripathi SC, Den Hollander P, Huang B, Lu M, et al. (2017). "Distinguishing mechanisms underlying EMT tristability". Cancer Convergence. 1 (1): 2. arXiv:1701.01746. Bibcode:2017arXiv170101746J. doi:10.1186/s41236-017-0005-8. PMC 5876698. PMID 29623961.
- Jolly MK, Tripathi SC, Jia D, Mooney SM, Celiktas M, Hanash SM, et al. (May 2016). "Stability of the hybrid epithelial/mesenchymal phenotype". Oncotarget. 7 (19): 27067–84. doi:10.18632/oncotarget.8166. PMC 5053633. PMID 27008704.