Comparative genomics
Comparative genomics is a field of biological research in which the genomic features of different organisms are compared.[2][3] The genomic features may include the DNA sequence, genes, gene order, regulatory sequences, and other genomic structural landmarks.[3] In this branch of genomics, whole or large parts of genomes resulting from genome projects are compared to study basic biological similarities and differences as well as evolutionary relationships between organisms.[2][4][5] The major principle of comparative genomics is that common features of two organisms will often be encoded within the DNA that is evolutionarily conserved between them.[6] Therefore, comparative genomic approaches start with making some form of alignment of genome sequences and looking for orthologous sequences (sequences that share a common ancestry) in the aligned genomes and checking to what extent those sequences are conserved. Based on these, genome and molecular evolution are inferred and this may in turn be put in the context of, for example, phenotypic evolution or population genetics.[7]

Virtually started as soon as the whole genomes of two organisms became available (that is, the genomes of the bacteria Haemophilus influenzae and Mycoplasma genitalium) in 1995, comparative genomics is now a standard component of the analysis of every new genome sequence.[2][8] With the explosion in the number of genome projects due to the advancements in DNA sequencing technologies, particularly the next-generation sequencing methods in late 2000s, this field has become more sophisticated, making it possible to deal with many genomes in a single study.[9] Comparative genomics has revealed high levels of similarity between closely related organisms, such as humans and chimpanzees, and, more surprisingly, similarity between seemingly distantly related organisms, such as humans and the yeast Saccharomyces cerevisiae.[4] It has also showed the extreme diversity of the gene composition in different evolutionary lineages.[8]
History
See also: History of genomics
Comparative genomics has a root in the comparison of virus genomes in the early 1980s.[8] For example, small RNA viruses infecting animals (picornaviruses) and those infecting plants (cowpea mosaic virus) were compared and turned out to share significant sequence similarity and, in part, the order of their genes.[10] In 1986, the first comparative genomic study at a larger scale was published, comparing the genomes of varicella-zoster virus and Epstein-Barr virus that contained more than 100 genes each.[11]
The first complete genome sequence of a cellular organism, that of Haemophilus influenzae Rd, was published in 1995.[12] The second genome sequencing paper was of the small parasitic bacterium Mycoplasma genitalium published in the same year.[13] Starting from this paper, reports on new genomes inevitably became comparative-genomic studies.[8]
Microbial genomes. The first high-resolution whole genome comparison system of microbial genomes of 10-15kbp was developed in 1998 by Art Delcher, Simon Kasif and Steven Salzberg and applied to the comparison of entire highly related microbial organisms with their collaborators at the Institute for Genomic Research (TIGR). The system is called MUMMER and was described in a publication in Nucleic Acids Research in 1999. The system helps researchers to identify large rearrangements, single base mutations, reversals, tandem repeat expansions and other polymorphisms. In bacteria, MUMMER enables the identification of polymorphisms that are responsible for virulence, pathogenicity, and anti-biotic resistance. The system was also applied to the Minimal Organism Project at TIGR and subsequently to many other comparative genomics projects.
Eukaryote genomes. Saccharomyces cerevisiae, the baker's yeast, was the first eukaryote to have its complete genome sequence published in 1996.[14] After the publication of the roundworm Caenorhabditis elegans genome in 1998[15] and together with the fruit fly Drosophila melanogaster genome in 2000,[16] Gerald M. Rubin and his team published a paper titled "Comparative Genomics of the Eukaryotes", in which they compared the genomes of the eukaryotes D. melanogaster, C. elegans, and S. cerevisiae, as well as the prokaryote H. influenzae.[17] At the same time, Bonnie Berger, Eric Lander, and their team published a paper on whole-genome comparison of human and mouse.[18]
With the publication of the large genomes of vertebrates in the 2000s, including human, the Japanese pufferfish Takifugu rubripes, and mouse, precomputed results of large genome comparisons have been released for downloading or for visualization in a genome browser. Instead of undertaking their own analyses, most biologists can access these large cross-species comparisons and avoid the impracticality caused by the size of the genomes.[19]
Next-generation sequencing methods, which were first introduced in 2007, have produced an enormous amount of genomic data and have allowed researchers to generate multiple (prokaryotic) draft genome sequences at once. These methods can also quickly uncover single-nucleotide polymorphisms, insertions and deletions by mapping unassembled reads against a well annotated reference genome, and thus provide a list of possible gene differences that may be the basis for any functional variation among strains.[9]
Evolutionary principles
One character of biology is evolution, evolutionary theory is also the theoretical foundation of comparative genomics, and at the same time the results of comparative genomics unprecedentedly enriched and developed the theory of evolution. When two or more of the genome sequence are compared, one can deduce the evolutionary relationships of the sequences in a phylogenetic tree. Based on a variety of biological genome data and the study of vertical and horizontal evolution processes, one can understand vital parts of the gene structure and its regulatory function.
Similarity of related genomes is the basis of comparative genomics. If two creatures have a recent common ancestor, the differences between the two species genomes are evolved from the ancestors' genome. The closer the relationship between two organisms, the higher the similarities between their genomes. If there is close relationship between them, then their genome will display a linear behaviour (synteny), namely some or all of the genetic sequences are conserved. Thus, the genome sequences can be used to identify gene function, by analyzing their homology (sequence similarity) to genes of known function.
Orthologous sequences are related sequences in different species: a gene exists in the original species, the species divided into two species, so genes in new species are orthologous to the sequence in the original species. Paralogous sequences are separated by gene cloning (gene duplication): if a particular gene in the genome is copied, then the copy of the two sequences is paralogous to the original gene. A pair of orthologous sequences is called orthologous pairs (orthologs), a pair of paralogous sequence is called collateral pairs (paralogs). Orthologous pairs usually have the same or similar function, which is not necessarily the case for collateral pairs. In collateral pairs, the sequences tend to evolve into having different functions.
Comparative genomics exploits both similarities and differences in the proteins, RNA, and regulatory regions of different organisms to infer how selection has acted upon these elements. Those elements that are responsible for similarities between different species should be conserved through time (stabilizing selection), while those elements responsible for differences among species should be divergent (positive selection). Finally, those elements that are unimportant to the evolutionary success of the organism will be unconserved (selection is neutral).
One of the important goals of the field is the identification of the mechanisms of eukaryotic genome evolution. It is however often complicated by the multiplicity of events that have taken place throughout the history of individual lineages, leaving only distorted and superimposed traces in the genome of each living organism. For this reason comparative genomics studies of small model organisms (for example the model Caenorhabditis elegans and closely related Caenorhabditis briggsae) are of great importance to advance our understanding of general mechanisms of evolution.[20][21]
Methods
Computational approaches are necessary for genome comparisons, given the large amount of data encoded in genomes. Many tools are now publicly available, ranging from whole genome comparisons to gene expression analysis.[22] This includes approaches from systems and control, information theory, string analysis and data mining.[23] Computational approaches will remain critical for research and teaching, especially when information science and genome biology is taught in conjunction.[24]
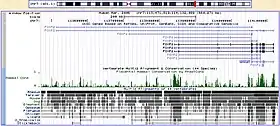
Comparative genomics starts with basic comparisons of genome size and gene density. For instance, genome size is important for coding capacity and possibly for regulatory reasons. High gene density facilitates genome annotation, analysis of environmental selection. By contrast, low gene density hampers the mapping of genetic disease as in the human genome.
Sequence alignment
Alignments are used to capture information about similar sequences such as ancestry, common evolutionary descent, or common structure and function. Alignments can be done for both genetic and protein sequences.[26][27] Alignments consist of local or global pairwise alignments, and multiple sequence alignments. One way to find global alignments is to use a dynamic programming algorithm known as Needleman-Wunsch algorithm. This algorithm can be modified and used to find local alignments.
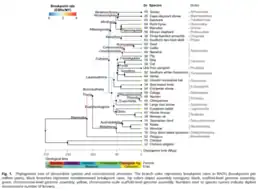
Phylogenetic reconstruction
Another computational method for comparative genomics is phylogenetic reconstruction. It is used to describe evolutionary relationships in terms of common ancestors. The relationships are usually represented in a tree called a phylogenetic tree. Similarly, coalescent theory is a retrospective model to trace alleles of a gene in a population to a single ancestral copy shared by members of the population. This is also known as the most recent common ancestor. Analysis based on coalescence theory tries predicting the amount of time between the introduction of a mutation and a particular allele or gene distribution in a population. This time period is equal to how long ago the most recent common ancestor existed. The inheritance relationships are visualized in a form similar to a phylogenetic tree. Coalescence (or the gene genealogy) can be visualized using dendrograms.[28]
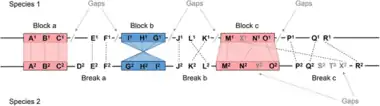
Genome maps
An additional method in comparative genomics is genetic mapping. In genetic mapping, visualizing synteny is one way to see the preserved order of genes on chromosomes. It is usually used for chromosomes of related species, both of which result from a common ancestor.[30] This and other methods can shed light on evolutionary history. A recent study used comparative genomics to reconstruct 16 ancestral karyotypes across the mammalian phylogeny. The computational reconstruction showed how chromosomes rearranged themselves during mammal evolution. It gave insight into conservation of select regions often associated with the control of developmental processes. In addition, it helped to provide an understanding of chromosome evolution and genetic diseases associated with DNA rearrangements.
Tools
Computational tools for analyzing sequences and complete genomes are developing quickly due to the availability of large amount of genomic data. At the same time, comparative analysis tools are progressed and improved. In the challenges about these analyses, it is very important to visualize the comparative results.[32]
Visualization of sequence conservation is a tough task of comparative sequence analysis. As we know, it is highly inefficient to examine the alignment of long genomic regions manually. Internet-based genome browsers provide many useful tools for investigating genomic sequences due to integrating all sequence-based biological information on genomic regions. When we extract large amount of relevant biological data, they can be very easy to use and less time-consuming.[32]
- UCSC Browser: This site contains the reference sequence and working draft assemblies for a large collection of genomes.[33]
- Ensembl: The Ensembl project produces genome databases for vertebrates and other eukaryotic species, and makes this information freely available online.[34]
- MapView: The Map Viewer provides a wide variety of genome mapping and sequencing data.[35]
- VISTA is a comprehensive suite of programs and databases for comparative analysis of genomic sequences. It was built to visualize the results of comparative analysis based on DNA alignments. The presentation of comparative data generated by VISTA can easily suit both small and large scale of data.[36]
- BlueJay Genome Browser: a stand-alone visualization tool for the multi-scale viewing of annotated genomes and other genomic elements.[37]
An advantage of using online tools is that these websites are being developed and updated constantly. There are many new settings and content can be used online to improve efficiency.[32]
Selected applications
Agriculture
Agriculture is a field that reaps the benefits of comparative genomics. Identifying the loci of advantageous genes is a key step in breeding crops that are optimized for greater yield, cost-efficiency, quality, and disease resistance. For example, one genome wide association study conducted on 517 rice landraces revealed 80 loci associated with several categories of agronomic performance, such as grain weight, amylose content, and drought tolerance. Many of the loci were previously uncharacterized.[38] Not only is this methodology powerful, it is also quick. Previous methods of identifying loci associated with agronomic performance required several generations of carefully monitored breeding of parent strains, a time-consuming effort that is unnecessary for comparative genomic studies.[39]
Vaccine development
The medical field also benefits from the study of comparative genomics. In an approach known as reverse vaccinology, researchers can discover candidate antigens for vaccine development by analyzing the genome of a pathogen or a family of pathogens.[40] Applying a comparative genomics approach by analyzing the genomes of several related pathogens can lead to the development of vaccines that are multi-protective. A team of researchers employed such an approach to create a universal vaccine for Group B Streptococcus, a group of bacteria responsible for severe neonatal infection.[41] Comparative genomics can also be used to generate specificity for vaccines against pathogens that are closely related to commensal microorganisms. For example, researchers used comparative genomic analysis of commensal and pathogenic strains of E. coli to identify pathogen-specific genes as a basis for finding antigens that result in immune response against pathogenic strains but not commensal ones.[42] In May 2019, using the Global Genome Set, a team in the UK and Australia sequenced thousands of globally-collected isolates of Group A Streptococcus, providing potential targets for developing a vaccine against the pathogen, also known as S. pyogenes.[43]
.png.webp)
Mouse models in immunology
T cells (also known as a T lymphocytes or a thymocytes) are immune cells that grow from stem cells in the bone marrow. They assist to defend the body from infection and may aid in the fight against cancer. Because of their morphological, physiological, and genetic resemblance to humans, mice and rats have long been the preferred species for biomedical research animal models. Comparative Medicine Research is built on the ability to use information from one species to understand the same processes in another. We can get new insights into molecular pathways by comparing human and mouse T cells and their effects on the immune system utilizing comparative genomics. In order to comprehend its TCRs and their genes, Glusman conducted research on the sequencing of the human and mouse T cell receptor loci. TCR genes are well-known and serve as a significant resource for supporting functional genomics and understanding how genes and intergenic regions of the genome contribute to biological processes.[44]
T-cell immune receptors are important in seeing the world of pathogens in the cellular immune system. One of the reasons for sequencing the human and mouse TCR loci was to match the orthologous gene family sequences and discover conserved areas using comparative genomics. These, it was thought, would reflect two sorts of biological information: (1) exons and (2) regulatory sequences. In fact, the majority of V, D, J, and C exons could be identified in this method. The variable regions are encoded by multiple unique DNA elements that are rearranged and connected during T cell (TCR) differentiation: variable (V), diversity (D), and joining (J) elements for the and polypeptides; and V and J elements for the and polypeptides.[Figure 1] However, several short noncoding conserved blocks of the genome had been shown. Both human and mouse motifs are largely clustered in the 200 bp [Figure 2], the known 3′ enhancers in the TCR/ were identified, and a conserved region of 100 bp in the mouse J intron was subsequently shown to have a regulatory function.
_(2).png.webp)
Comparisons of the genomic sequences within each physical site or location of a specific gene on a chromosome (locs) and across species allow for research on other mechanisms and other regulatory signals. Some suggest new hypotheses about the evolution of TCRs, to be tested (and improved) by comparison to the TCR gene complement of other vertebrate species. A comparative genomic investigation of humans and mice will obviously allow for the discovery and annotation of many other genes, as well as identifying in other species for regulatory sequences.[44]
Research
Comparative genomics also opens up new avenues in other areas of research. As DNA sequencing technology has become more accessible, the number of sequenced genomes has grown. With the increasing reservoir of available genomic data, the potency of comparative genomic inference has grown as well.
A notable case of this increased potency is found in recent primate research. Comparative genomic methods have allowed researchers to gather information about genetic variation, differential gene expression, and evolutionary dynamics in primates that were indiscernible using previous data and methods.[45]
Great Ape Genome Project
The Great Ape Genome Project used comparative genomic methods to investigate genetic variation with reference to the six great ape species, finding healthy levels of variation in their gene pool despite shrinking population size.[46] Another study showed that patterns of DNA methylation, which are a known regulation mechanism for gene expression, differ in the prefrontal cortex of humans versus chimps, and implicated this difference in the evolutionary divergence of the two species.[47]
See also
References
- Darling AE, Miklós I, Ragan MA (July 2008). "Dynamics of genome rearrangement in bacterial populations". PLOS Genetics. 4 (7): e1000128. doi:10.1371/journal.pgen.1000128. PMC 2483231. PMID 18650965.

- Touchman J (2010). "Comparative Genomics". Nature Education Knowledge. 3 (10): 13.
- Xia X (2013). Comparative Genomics. SpringerBriefs in Genetics. Heidelberg: Springer. doi:10.1007/978-3-642-37146-2. ISBN 978-3-642-37145-5. S2CID 5491782.
- Russel PJ, Hertz PE, McMillan B (2011). Biology: The Dynamic Science (2nd ed.). Belmont, CA: Brooks/Cole. pp. 409–410.
- Primrose SB, Twyman RM (2003). Principles of Genome Analysis and Genomics (3rd ed.). Malden, MA: Blackwell Publishing. ISBN 9781405101202.
- Hardison RC (November 2003). "Comparative genomics". PLOS Biology. 1 (2): E58. doi:10.1371/journal.pbio.0000058. PMC 261895. PMID 14624258.
- Ellegren H (November 2008). "Comparative genomics and the study of evolution by natural selection". Molecular Ecology. 17 (21): 4586–4596. doi:10.1111/j.1365-294X.2008.03954.x. PMID 19140982. S2CID 43171654.
- Koonin EV, Galperin MY (2003). Sequence - Evolution - Function: Computational approaches in comparative genomics. Dordrecht: Springer Science+Business Media.
- Hu B, Xie G, Lo CC, Starkenburg SR, Chain PS (November 2011). "Pathogen comparative genomics in the next-generation sequencing era: genome alignments, pangenomics and metagenomics". Briefings in Functional Genomics. 10 (6): 322–333. doi:10.1093/bfgp/elr042. PMID 22199376.
- Argos P, Kamer G, Nicklin MJ, Wimmer E (September 1984). "Similarity in gene organization and homology between proteins of animal picornaviruses and a plant comovirus suggest common ancestry of these virus families". Nucleic Acids Research. 12 (18): 7251–7267. doi:10.1093/nar/12.18.7251. PMC 320155. PMID 6384934.
- McGeoch DJ, Davison AJ (May 1986). "DNA sequence of the herpes simplex virus type 1 gene encoding glycoprotein gH, and identification of homologues in the genomes of varicella-zoster virus and Epstein-Barr virus". Nucleic Acids Research. 14 (10): 4281–4292. doi:10.1093/nar/14.10.4281. PMC 339861. PMID 3012465.
- Fleischmann RD, Adams MD, White O, Clayton RA, Kirkness EF, Kerlavage AR, et al. (July 1995). "Whole-genome random sequencing and assembly of Haemophilus influenzae Rd". Science. 269 (5223): 496–512. Bibcode:1995Sci...269..496F. doi:10.1126/science.7542800. PMID 7542800.
- Fraser CM, Gocayne JD, White O, Adams MD, Clayton RA, Fleischmann RD, et al. (October 1995). "The minimal gene complement of Mycoplasma genitalium". Science. 270 (5235): 397–403. Bibcode:1995Sci...270..397F. doi:10.1126/science.270.5235.397. PMID 7569993. S2CID 29825758.
- Goffeau A, Barrell BG, Bussey H, Davis RW, Dujon B, Feldmann H, et al. (October 1996). "Life with 6000 genes". Science. 274 (5287): 546, 563–546, 567. Bibcode:1996Sci...274..546G. doi:10.1126/science.274.5287.546. PMID 8849441. S2CID 16763139.
- The C. elegans Sequencing Consortium (December 1998). "Genome sequence of the nematode C. elegans: a platform for investigating biology". Science. 282 (5396): 2012–2018. Bibcode:1998Sci...282.2012.. doi:10.1126/science.282.5396.2012. PMID 9851916.
- Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG, et al. (March 2000). "The genome sequence of Drosophila melanogaster". Science. 287 (5461): 2185–2195. Bibcode:2000Sci...287.2185.. CiteSeerX 10.1.1.549.8639. doi:10.1126/science.287.5461.2185. PMID 10731132.
- Rubin GM, Yandell MD, Wortman JR, Gabor Miklos GL, Nelson CR, Hariharan IK, et al. (March 2000). "Comparative genomics of the eukaryotes". Science. 287 (5461): 2204–2215. Bibcode:2000Sci...287.2204.. doi:10.1126/science.287.5461.2204. PMC 2754258. PMID 10731134.
- Batzoglou S, Pachter L, Mesirov JP, Berger B, Lander ES (July 2000). "Human and mouse gene structure: comparative analysis and application to exon prediction". Genome Research. 10 (7): 950–958. doi:10.1101/gr.10.7.950. PMC 310911. PMID 10899144.
- Ureta-Vidal A, Ettwiller L, Birney E (April 2003). "Comparative genomics: genome-wide analysis in metazoan eukaryotes". Nature Reviews. Genetics. 4 (4): 251–262. doi:10.1038/nrg1043. PMID 12671656. S2CID 2037634.
- Stein LD, Bao Z, Blasiar D, Blumenthal T, Brent MR, Chen N, et al. (November 2003). "The genome sequence of Caenorhabditis briggsae: a platform for comparative genomics". PLOS Biology. 1 (2): E45. doi:10.1371/journal.pbio.0000045. PMC 261899. PMID 14624247.
- "Newly Sequenced Worm a Boon for Worm Biologists". PLOS Biology. 1 (2): e4. 2003. doi:10.1371/journal.pbio.0000044.
- Cristianini N, Hahn M (2006). Introduction to Computational Genomics. Cambridge University Press. ISBN 978-0-521-67191-0.
- Pratas D, Silva RM, Pinho AJ, Ferreira PJ (May 2015). "An alignment-free method to find and visualise rearrangements between pairs of DNA sequences". Scientific Reports. 5: 10203. Bibcode:2015NatSR...510203P. doi:10.1038/srep10203. PMC 4434998. PMID 25984837.
- Via A, De Las Rivas J, Attwood TK, Landsman D, Brazas MD, Leunissen JA, et al. (October 2011). "Ten simple rules for developing a short bioinformatics training course". PLOS Computational Biology. 7 (10): e1002245. Bibcode:2011PLSCB...7E2245V. doi:10.1371/journal.pcbi.1002245. PMC 3203054. PMID 22046119.
- Damas J, Corbo M, Kim J, Turner-Maier J, Farré M, Larkin DM, et al. (October 2022). "Evolution of the ancestral mammalian karyotype and syntenic regions". Proceedings of the National Academy of Sciences of the United States of America. 119 (40): e2209139119. doi:10.1073/pnas.2209139119. PMC 9550189. PMID 36161960.
- Altschul SF, Pop M (2017). "Sequence Alignment". In Rosen KH, Shier DR, Goddard W (eds.). Handbook of Discrete and Combinatorial Mathematics (2nd ed.). Boca Raton (FL): CRC Press/Taylor & Francis. ISBN 978-1-58488-780-5. PMID 29206392. Retrieved 2022-12-18.
- Prjibelski AD, Korobeynikov AI, Lapidus AL (2019-01-01). "Sequence Analysis". In Ranganathan S, Gribskov M, Nakai K, Schönbach C (eds.). Encyclopedia of Bioinformatics and Computational Biology. Oxford: Academic Press. pp. 292–322. doi:10.1016/b978-0-12-809633-8.20106-4. ISBN 978-0-12-811432-2. S2CID 226247797.
- Haubold B, Wiehe T (September 2004). "Comparative genomics: methods and applications". Die Naturwissenschaften. 91 (9): 405–421. doi:10.1007/s00114-004-0542-8. PMID 15278216. S2CID 2041895.
- Liu D, Hunt M, Tsai IJ (January 2018). "Inferring synteny between genome assemblies: a systematic evaluation". BMC Bioinformatics. 19 (1): 26. doi:10.1186/s12859-018-2026-4. PMC 5791376. PMID 29382321.
- Duran C, Edwards D, Batley J (2009). "Genetic maps and the use of synteny". Plant Genomics. Methods in Molecular Biology. Vol. 513. pp. 41–55. doi:10.1007/978-1-59745-427-8_3. ISBN 978-1-58829-997-0. PMID 19347649.
- Damas J, Corbo M, Kim J, Turner-Maier J, Farré M, Larkin DM, et al. (October 2022). "Evolution of the ancestral mammalian karyotype and syntenic regions". Proceedings of the National Academy of Sciences of the United States of America. 119 (40): e2209139119. doi:10.1073/pnas.2209139119. PMC 9550189. PMID 36161960.
- Bergman NH (2007). Bergman NH (ed.). Comparative Genomics: Volumes 1 and 2. Totowa, New Jersey: Humana Press. ISBN 978-193411-537-4. PMID 21250292.
- "UCSC Browser".
- "Ensembl Genome Browser". Archived from the original on 2013-10-21.
- "Map Viewer".
- "VISTA tools".
- Soh J, Gordon PM, Sensen CW (March 2012). "The Bluejay genome browser". Current Protocols in Bioinformatics. John Wiley & Sons, Inc. 37: Unit10.9. doi:10.1002/0471250953.bi1009s37. ISBN 9780471250951. PMID 22389011. S2CID 34553139.
- Huang X, Wei X, Sang T, Zhao Q, Feng Q, Zhao Y, et al. (November 2010). "Genome-wide association studies of 14 agronomic traits in rice landraces". Nature Genetics. 42 (11): 961–967. doi:10.1038/ng.695. PMID 20972439. S2CID 439442.
- Morrell PL, Buckler ES, Ross-Ibarra J (December 2011). "Crop genomics: advances and applications". Nature Reviews. Genetics. 13 (2): 85–96. doi:10.1038/nrg3097. PMID 22207165. S2CID 13358998.
- Seib KL, Zhao X, Rappuoli R (October 2012). "Developing vaccines in the era of genomics: a decade of reverse vaccinology". Clinical Microbiology and Infection. 18 Suppl 5 (SI): 109–116. doi:10.1111/j.1469-0691.2012.03939.x. PMID 22882709.
- Maione D, Margarit I, Rinaudo CD, Masignani V, Mora M, Scarselli M, et al. (July 2005). "Identification of a universal Group B streptococcus vaccine by multiple genome screen". Science. 309 (5731): 148–150. Bibcode:2005Sci...309..148M. doi:10.1126/science.1109869. PMC 1351092. PMID 15994562.
- Rasko DA, Rosovitz MJ, Myers GS, Mongodin EF, Fricke WF, Gajer P, et al. (October 2008). "The pangenome structure of Escherichia coli: comparative genomic analysis of E. coli commensal and pathogenic isolates". Journal of Bacteriology. 190 (20): 6881–6893. doi:10.1128/JB.00619-08. PMC 2566221. PMID 18676672.
- "Group a Streptococcus Vaccine Target Candidates Identified from Global Genome Set". 28 May 2019.
- Glusman G, Rowen L, Lee I, Boysen C, Roach JC, Smit AF, et al. (September 2001). "Comparative genomics of the human and mouse T cell receptor loci". Immunity. 15 (3): 337–349. doi:10.1016/s1074-7613(01)00200-x. PMID 11567625.
- Rogers J, Gibbs RA (May 2014). "Comparative primate genomics: emerging patterns of genome content and dynamics". Nature Reviews. Genetics. 15 (5): 347–359. doi:10.1038/nrg3707. PMC 4113315. PMID 24709753.
- Prado-Martinez J, Sudmant PH, Kidd JM, Li H, Kelley JL, Lorente-Galdos B, et al. (July 2013). "Great ape genetic diversity and population history". Nature. 499 (7459): 471–475. Bibcode:2013Natur.499..471P. doi:10.1038/nature12228. PMC 3822165. PMID 23823723.
- Zeng J, Konopka G, Hunt BG, Preuss TM, Geschwind D, Yi SV (September 2012). "Divergent whole-genome methylation maps of human and chimpanzee brains reveal epigenetic basis of human regulatory evolution". American Journal of Human Genetics. 91 (3): 455–465. doi:10.1016/j.ajhg.2012.07.024. PMC 3511995. PMID 22922032.
Further reading
- Kellis M, Patterson N, Endrizzi M, Birren B, Lander ES (May 2003). "Sequencing and comparison of yeast species to identify genes and regulatory elements". Nature. 423 (6937): 241–254. Bibcode:2003Natur.423..241K. doi:10.1038/nature01644. PMID 12748633. S2CID 1530261.
- Cliften P, Sudarsanam P, Desikan A, Fulton L, Fulton B, Majors J, et al. (July 2003). "Finding functional features in Saccharomyces genomes by phylogenetic footprinting". Science. 301 (5629): 71–76. Bibcode:2003Sci...301...71C. doi:10.1126/science.1084337. PMID 12775844. S2CID 1305166.
- Boffelli D, McAuliffe J, Ovcharenko D, Lewis KD, Ovcharenko I, Pachter L, Rubin EM (February 2003). "Phylogenetic shadowing of primate sequences to find functional regions of the human genome". Science. 299 (5611): 1391–1394. doi:10.1126/science.1081331. PMID 12610304. S2CID 17217612.
- Dujon B, Sherman D, Fischer G, Durrens P, Casaregola S, Lafontaine I, et al. (July 2004). "Genome evolution in yeasts". Nature. 430 (6995): 35–44. Bibcode:2004Natur.430...35D. doi:10.1038/nature02579. PMID 15229592. S2CID 4399964.
- Filipski A, Kumar S (2005). "Comparative genomics in eukaryotes". In Gregory TR (ed.). The Evolution of the Genome. San Diego: Elsevier. pp. 521–583.
- Gregory TR, DeSalle R (2005). "Comparative genomics in prokaryotes". In Gregory TR (ed.). The Evolution of the Genome. San Diego: Elsevier. pp. 585–675.
- Xie X, Lu J, Kulbokas EJ, Golub TR, Mootha V, Lindblad-Toh K, et al. (March 2005). "Systematic discovery of regulatory motifs in human promoters and 3' UTRs by comparison of several mammals". Nature. 434 (7031): 338–345. Bibcode:2005Natur.434..338X. doi:10.1038/nature03441. PMC 2923337. PMID 15735639.
- Champ PC, Binnewies TT, Nielsen N, Zinman G, Kiil K, Wu H, et al. (March 2006). "Genome update: purine strand bias in 280 bacterial genomes". Microbiology. 152 (Pt 3): 579–583. doi:10.1099/mic.0.28637-0. PMID 16514138.
- Kumar L, Breakspear A, Kistler C, Ma LJ, Xie X (March 2010). "Systematic discovery of regulatory motifs in Fusarium graminearum by comparing four Fusarium genomes". BMC Genomics. 11: 208. doi:10.1186/1471-2164-11-208. PMC 2853525. PMID 20346147.
- Batzoglou S, Pachter L, Mesirov JP, Berger B, Lander ES (July 2000). "Human and mouse gene structure: comparative analysis and application to exon prediction". Genome Research. 10 (7): 950–958. doi:10.1101/gr.10.7.950. PMC 310911. PMID 10899144.
External links
- Genomes OnLine Database (GOLD)
- Genome News Network
- JCVI Comprehensive Microbial Resource
- Pathema: A Clade Specific Bioinformatics Resource Center
- CBS Genome Atlas Database
- The UCSC Genome Browser
- The U.S. National Human Genome Research Institute
- Ensembl The Ensembl Genome Browser
- Genolevures, comparative genomics of the Hemiascomycetous yeasts
- Phylogenetically Inferred Groups (PhIGs), a recently developed method incorporates phylogenetic signals in building gene clusters for use in comparative genomics.
- Metazome Archived 2006-08-10 at the Wayback Machine, a resource for the phylogenomic exploration and analysis of Metazoan gene families.
- IMG The Integrated Microbial Genomes system, for comparative genome analysis by the DOE-JGI.
- Dcode.org Dcode.org Comparative Genomics Center.
- SUPERFAMILY Protein annotations for all completely sequenced organisms
- Comparative Genomics
- Blastology and Open Source: Needs and Deeds
- Alignment-free comparative Genomics tool
| Genomics | |
|---|---|
| Bioinformatics | |
| Structural biology | |
| Research tools | |
| Organizations | |
| |