Innate lymphoid cell
Innate lymphoid cells (ILCs) are the most recently discovered family of innate immune cells, derived from common lymphoid progenitors (CLPs). In response to pathogenic tissue damage, ILCs contribute to immunity via the secretion of signalling molecules, and the regulation of both innate and adaptive immune cells. ILCs are primarily tissue resident cells, found in both lymphoid (immune associated), and non- lymphoid tissues, and rarely in the blood. They are particularly abundant at mucosal surfaces, playing a key role in mucosal immunity and homeostasis. Characteristics allowing their differentiation from other immune cells include the regular lymphoid morphology, absence of rearranged antigen receptors found on T cells and B cells (due to the lack of the RAG gene), and phenotypic markers usually present on myeloid or dendritic cells.[1]
Based on the difference in developmental pathways, phenotype, and signalling molecules produced, in 2013, ILCs were divided into three groups: 1, 2 and 3, however, after further investigation, they are now divided into five groups: NK cells, ILC1s, ILC2s, ILC3s, and lymphoid tissue inducer (LTi) cells.[2] ILCs are implicated in multiple physiological functions, including tissue homeostasis, morphogenesis, metabolism, repair, and regeneration. Many of their roles are similar to T cells, therefore they have been suggested to be the innate counterparts of T cells.[3] The dysregulation of ILCs can lead to immune pathology such as allergy, bronchial asthma and autoimmune disease.[4]
Classification
The development of ILCs is initiated in response to the presence of transcription factors that are switched on due to the presence of surrounding microenvironmental factors, such as: cytokines, notch ligands, and circadian rhythm (inbuilt behavioural changes following a daily cycle). Once matured, the ILCs release cytokines. The classification of ILCs is therefore based on the differences in the transcription factor and cytokine profiles associated with the development and function of the different ILC subtypes.[5]
| Stimuli | Tissue Signals | Cell | Mediators | Immune Function |
|---|---|---|---|---|
| Tumours
Intracellular microbes (virus, bacteria, parasite) |
IL-12 |  |
IFN-γ | Type 1 immunity (Macrophage activation, cytotoxicity, oxygen radicals) |
| Large extracellular molecules (parasites and allergens) | IL-25 |  |
IL-4, IL-5, IL-13, IL-9 | Type 2 immunity (Mucus production, alternative macrophage activation, extracellular matrix/tissue repair, vasodilation, thermoregulation) |
| Extracellular microbes (bacteria, fungi) | IL-1B |  |
IL-22, IL-17 | Type 3 immunity (Phagocytosis, antimicrobial peptides, epithelium survival) |
| Mesenchymal organizer cells (retinoic acid, CXCL13, RANK-L) | IL-1B |  |
RANK, TNF, Lymphotoxin | Formation of secondary lymphoid structures |
Group 1 ILCs
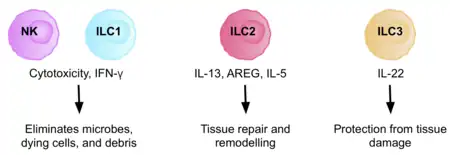
ILC1 and NK cell lineages diverge early in their developmental pathways and can be discriminated by their difference in dependence on transcription factors, their cytotoxicity, and their resident marker expression. NK cells are cytotoxic cells, circulating in the bloodstream, killing virus-infected, and tumor cells. ILC1s, are non- cytotoxic or weakly cytotoxic, tissue resident cells, functioning in the defence against infections with viruses and certain bacteria.
Due to ILC1s and NK cells having both shared and unshared features, the classification of human ILC1s has been problematic. Both cell types produce IFN-γ as their principle cytokine and require the transcription factor T-bet to do so.[6] Both cells can also produce IFN-γ when the cytokines IL-15 or IL-12 are up-regulated in tissues after infection or injury, and secrete TGFβ1 in tandem with IFN-γ when stimulated. This drives gut epithelial and extra-cellular matrix remodelling.[7] IL-18 co-stimulation also significantly increases IFN-γ levels.[8] The release of IFN-γ stimulates macrophages and other mononuclear phagocytes, to induce an antimicrobial effect to eradicate intracellular infections. Oxygen radicals produced by both cell types also aid in the eradication of infection. ILC1s and NK cells can also produce TNF- α, further contributing to the inflammatory response, depending on their molecule expression.
There are differences in dependence on transcription factors between NK cells and ILC1s. Although both cell types use T-bet for development, NK cells have been found to be present in T-bet deficient hosts, but ILC1s are completely dependent on its presence.[6] Development of NK cells is, however, completely dependent on the presence of the transcription factor Eomes, whereas ILC1s can develop independent of its presence.[6] This means, Eomes can generally be used as a marker for NK cells, suggesting that mature NK cells are Tbet + Eomes +, and ILC1 are Tbet + Eomes -.[9]
ILC1s and NK cells have some phenotypic markers in common, including: NK1.1 in mice, and NK cell receptors (NCRs) such as NKp44 and NKp46 in both humans and mice.[10][6] They also have differences in phenotypic markers, including the expression of CD127 on human ILC1s, which is not present on all NK cells. In addition, NKp80, a marker for human NK cells, is not expressed on ILC1s. In mice, CD200R has been shown to distinguish NK cells from ILC1s.[11] The relationship between the ILC1 and NK cell lineages still remains fuzzy due to a lack of these characteristic markers present on some NK/ILC1 cells in certain tissues, or after certain infection/inflammation events. This supports the tissue specific function theory.[10] For example, CD127, although expressed by the majority of ILC1s, is absent from the salivary gland resident ILC1s, which also have the ability to express Eomes, a fundamental feature of NK cells.[12]
Due to the production of granzymes and perforin, NK cells are considered the innate counterparts of cytotoxic CD8+ T cells, whereas ILC1s are considered the innate counterpart of T helper cells, due to the sole production of IFN-γ without cytotoxic activity.[13]
Group 2 ILCs
ILC2s are tissue resident and involved in the innate response to parasites, such as helminth infection, by helping repair tissue damage. They are abundant in tissues of the skin,[14][15] lung, liver, and gut.[6][16] They are characterised by the production of amphiregulin, and type 2 cytokines, including IL-4, IL-5, and IL-13, in response to IL-25, TSLP, and IL-33.[6] Due to their cytokine signature, they are considered the innate counterparts of Th2 cells.
They express characteristic surface markers and receptors for chemokines, which are involved in the distribution of lymphoid cells to specific organ sites. In humans, ILC2s express CRTH2, KLRG1, SST2, CD161, and CD25.[3] In mice, ILC2s express CD44, but not CD161.[3]
ILC2s require IL-7 for their development, activating the fundamental transcription factors RORα and GATA3. GATA3 is also required for maintenance of ILC2 function, with GATA3 deprivation inhibiting the development and function of the cells.
Although considered homogenous, ILC2s can be classified into subpopulations of natural ILC2s (nILC2s), and inflammatory ILC2s (iILC2s), dependent on their responsiveness to IL-33 and IL-25.[3] nILC2s are those responsive to IL-33 in tissues in a natural immune state, while iILC2s respond to IL-25 or the helminth parasite.[3] nILC2s express more Thy1 and ST2, and reduced KLRG1.[3] iILC2s, express more KLRG1, and reduced Thy1 and ST2.[3] In addition to these subpopulations, another population, named the ILC210 cell, is characterised by its ability to produce IL-10.[3]
Group 3 ILCs
ILC3s are involved in the innate immune response to extracellular bacteria and fungi. They play a key role in homeostasis of the intestinal bacteria and in regulating Th17 cell responses.[17] Human adult ILC3s, are primarily found in the lamina propria of the intestine, and the tonsils, however, they are also found in the spleen, endometrium, decidua, and skin.[18]
ILC3s are dependent on the transcription factor RORγt for their development and function.[19] They express RORγt in response to IL- 1β and IL-23, or pathogenic signals.[20] IL-22 is the principle cytokine produced by ILC3s and plays a fundamental role in maintaining intestinal homeostasis. However, ILC3s produce a variety of other cytokines, including IL-17, IL-22, IFN- γ, and GM-CSF, depending on the environmental stimuli.[21]
There are two subsets of ILC3s, NCR- and NCR+ ILC3s, with the displayed NCR on mice ILC3s being NKp46, in comparison to NKp44 displayed on human ILC3s.[21] NKp44+ ILC3s are highly enriched in the tonsils and intestines, as an exclusive source of IL-22.[21] Some ILC3s can also express other NK cell markers, including NKp30 and CD56.[22] NCR- ILC3s mainly produce IL-17A and IL-17F, and under certain circumstances, IL-22.[23] NCR- ILC3s can differentiate into NCR+ upon increased expression levels of T-bet.[5] Despite expressing NK cell markers, ILC3s differ greatly from NK cells, with different developmental pathways and effector functions.
Lymphoid Tissue inducer (LTi) cells
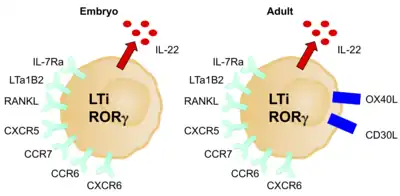
LTi cells are considered a separate lineage due to their unique developmental pathway, however, they are often considered to be part of the ILC3 group due to their many similar characteristics. Like ILC3s, LTi cells are dependent on RORγt. They are involved in the formation of secondary lymph nodes and Peyer’s patches by promoting lymphoid tissue development, which they do through the action of lymphotoxin, a member of the TNF superfamily.[6] They are critical during both the embryonic and adult stages of development of the immune system, and therefore LTi cells are present in organs and tissues early during embryonal development.[6] They have a pivotal role in primary and secondary lymphoid tissue organisation and in adult lymphoid tissue, regulating the adaptive immune response and maintaining secondary lymphoid tissue structures.[25]
Their production is stimulated by retinoic acid, CXCL13, RANK-L, and the cytokines IL-1B, IL-23, and IL-6.[26] They express c- Kit, CCR6, CD25, CD127, and CD90, however, no NCRs.[6] The expression of OX40L is another good marker for LTi cells in adult mice and humans.[24] They can be either CD4+/-. Like ILC3s, upon activation, LTi cells mostly produce IL-17A, IL-17F, and IL-22.[23] They are mediated by RANK, TNF, IL-17, and IL-22.
LTi cells induce the expression of AIRE, the autoimmune regulatory gene, by allowing development of embryonic thymic epithelial cells.[24] They do this via lymphotoxin α4β7 and RANK-L signalling.[24] LTi cells also allow the survival of memory CD4+ T cells, and therefore memory immune responses, within newly formed lymph nodes.[24] They do this via the TNF superfamily members OX40L and CD30L, which signal to CD4+ T cells.[24] This role could be used to prevent autoimmunity and to enhance memory responses after vaccination.[24]
Development
Our understanding of the pathways involved in the development of ILCs has only become clear in the last few years, with our knowledge mainly based on mouse pathways.[6] CLPs have the ability to differentiate into a number of different cell types including T cells, B cells, and ILCs, depending on the cellular signals present. With the exception of NK cells, all ILCs require IL-7 signalling for survival. The transcriptional repressor ID2 appears to antagonize B and T cell differentiation, yielding an ID2-dependent precursor that can further differentiate with lineage-specific transcription factors.[4]
ILCs are recombination activating gene (RAG)- independent, instead, they rely on cytokine signalling through the common cytokine- receptor gamma chain and the JAK3 kinase pathway for development.[27]
Early Development
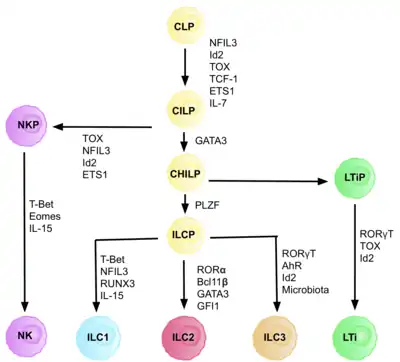
ILCs are derived from common innate lymphoid progenitors (CILPs), which are derived from CLPs, which have the ability to differentiate into a number of different lymphoid cell types including T and B cells.[6] CILPs can then differentiate into NK cell precursors (NKP), or the more recently described common helper innate lymphoid progenitors (CHILPs).[6] CHILPs can then differentiate into lymphoid tissue inducer progenitors (LTiPs), and innate lymphoid cell precursors (ILCPs). The factors present in the microenvironment determine the progression of CLPs towards specific ILC subtypes, including notch ligands, cytokines, circadian rhythm, and the expression of transcription factors.
Identification of the ILC progenitor cell (ILCP)
The development of CLPs to CILPs and on to ILCs requires the transcription factor ID2, to mediate suppression of the lymphoid cell fates generating T and B cells.[27] It does this via reducing activity of E-box transcription factors (E2A, E2-2, and HEB), critical in B and T cell development.[27] Initially it was assumed that ID2 was required in order for CLPs to differentiate into all ILC subsets, however, research showed that knock out of ID2 during CLP development, cripples the development of all ILC subsets other than NK cell progenitors, which are not reliant on the presence of Id2.[28] Due to this realisation, a group of lineage negative cells (requirement of any true precursor cell), that were entirely dependent on the presence of ID2, and expressed other key ILC markers, were identified, with the phenotype: Lin-ID2+IL7Ra+CD25-α4β7+, which are now known as the common helper like innate lymphoid progenitors CHILPs.[28] They are named ‘common helper like’ due to their similarity to the T helper effector cell fates.
Transcription factor dependence
Each stage of differentiation is dependent on expression of different transcription factors, including: NFIL3, TCF-1, ETS1, GATA3, PLZF, T-bet, Eomes, RUNX3, RORα, Bcl11b, Gfi1, RORγt, and AhR.[6] The coordinated expression of these specific transcription factors activate or repress target genes critical in the differentiation of the lymphocyte subsets.[27] In particular, Nfil3, whose expression is regulated by cytokines, controls the differentiation of ILCs via the transcription factors Id2, RORγt, Eomes, and Tox.[29] This provides evidence for the tissue signals playing a key role in fate decisions into ILC lineages.
Origin and migration
Studies suggest primary site of ILC development is in the liver in the foetus, and the bone marrow in adults, as this is where CLPs, NKPs, and CHILPs have been found.[27] The cells then exit and circulate in the blood until they reach their designated tissues, coded for by adhesion molecules and chemokines.[27] However, it has also been shown that the maturation of the ILCs can take place outside the primary lymphoid tissues, similar to the maturation of naïve T helper cells.
NK cell precursors, and ILC3 precursors have been found in the human tonsil, and foetal ILCPs present in the mouse intestine, accumulating in the Peyer’s Patches.[30][31] Retinoic acid, produced by many cell types, such as nerve cells, dendritic cells, and stromal cells, favours the differentiation of ILC3s, rather than ILC2s, and it is required for their complete maturation.[27] In addition, AhR, which can be triggered through ligands produced after the catabolism food, is required for the maintenance of function and expression of intestinal ILC3s.[30]
Function
ILCs participate in our immune response to pathogens in all organs, in particular at mucosal surfaces.[13] They are key in the innate immune response due to their ability to rapidly secrete immunoregulatory cytokines, however, they also play a role in the shaping of the adaptive response by interacting with other immune cells. The microenvironment of the tissue they reside in determines and fine- tunes the expression of the diverse ILC profiles, facilitating their interaction in multiple effector functions.
The strategic positioning and deep rooting of ILCs within tissues allow them to maintain homeostasis, and therefore healthy tissue functioning. However, the ILCs also have detrimental roles in different mucosal sites.[32]
Since the function of ILCs is linked to their specific tissue localization, determination of the signals involved in their localization and migration patterns will be important in the identification of new avenues for treatment of diseases.[21]
Helminth infection and tissue repair
A fundamental property of type 2 immunity, and therefore ILC2 cells, is to deal with oversized organisms, that cannot be digested, such as the helminths.[33] In the intestine, in response to a helminth infection, epithelial cells secrete high levels of IL-25, activating ILC2 cells. ILC2s produce IL-13, which drives the differentiation of additional epithelial cells, via Notch signalling pathways. This instruction allows the tissue to be remodelled to allow for the expulsion of the helminth parasite, and other large pathogens.
IL-13 also activates T cells, inducing further physiological responses to expel the parasite.[34] T cells stimulate goblet cell mucus secretion, contraction of smooth muscle, and they secrete signals recruiting mast cells and eosinophils to the site, stimulating B cell proliferation.[34]
The infection can lead to tissue damage, due to migration of the helminth. ILC2s have a key role in repairing the tissue damage after infection, by producing ligands such as AREG, for epithelial growth factor receptors, which facilitates differentiation of epithelial cells for tissue repair.[6] This can function to enhance the barrier function of the epithelium and slow pathogen entry.[34]
In multiple tissue niches, ILCs have a relationship with non- hematopoietic cells such as stromal cells. In the lung, ILC2s have a distinct localization to stromal cells, which release IL-33, and TSLP, promoting ILC2 homeostasis, in both the steady state, and in response to helminth infection, after the helminth has developed in the intestine, and migrated to the lung through the blood.[35]
Lung ILC2s are positioned close to blood vessels, to allow recruitment of eosinophils from the blood. They are also positioned within the airways, where potential pathogens may accumulate. This means they are in close contact with neuroendocrine cells, which activate ILC2s via the release of calcitonin gene-related peptide.[36] Other studies also confirm the regulation of ILC function via neuronal circuits.
In addition, ILC1s and ILC3s release oxygen radicals and lethally damaging enzymes in response to pathogenic infection, causing damage to the host tissue. The repair responses for the tissue are coordinated by the type 2 immune response, after the ILC3s and ILC1s have cleansed the tissue of microbes and debris.
Intestinal mucosa
Intestinal ILCs are exposed to dietary, microbial, and endogenous metabolites. ILC homing to the small intestine is mediated by α4β7 integrin, and the receptor CCR9. ILC2s express CCR9 in the bone marrow, so can directly home to the intestine, however, retinoic acid is required to allow CCR9 expression on ILC1s, and ILC3s.
ILCs facilitate maintenance of barrier integrity in the intestine, protecting from various bacteria and viral infections. ILC3s are the most abundant subset present in both the adult and foetal intestine.[37] The distribution of ILCs in the intestine changes during development, and they are unevenly distributed throughout the segments of the gastro-intestinal tract. This distribution to different niches within the intestine is mediated through distinct signalling cascades.[38] In humans, approximately 70% of the intestinal ILCs are NCR+, and 15% are NCR-.[39]
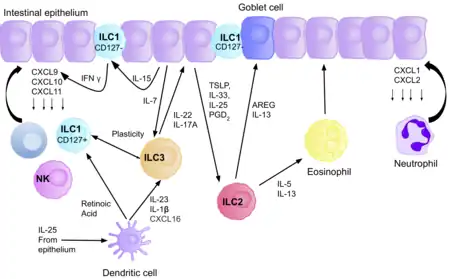
ILC3s directly interact with bacterial flora, creating a network between the microbiota, and the host, favouring homeostasis. ILC3s restrict colonization of multiple unbeneficial bacteria in the gut, via secretion of IL-22, stimulating epithelial cells to produce antimicrobial peptides.[40] The IL-22 production is induced due to the production of IL-23 and IL-1β by macrophages and DCs, and it promotes mucosal layer healing.[3] For example, IL-22 can promote repair of intestinal damage after chemotherapy or radiotherapy. ILC3s regulate the containment of commensal bacteria in the lumen, allowing it to be exposed to lamina propria phagocytes, leading to T cell priming. Although they can present antigens, via MHC class II receptors, ILCs lack co-stimulatory molecules, and therefore play a role in T cell anergy, promoting tolerance to beneficial commensals.[39] The relationship between ILC3s, and T cells in the gut is therefore crucial for maintaining homeostasis, as in the absence of ILC3s, there could be uncontrolled T cell activation. In addition, microbiota play a role in fine tuning IL-22 production by ILC3s, for example, segmented filamentous bacteria in the ileum regulate IL-22 production and allow differentiation of Th17 cells.[41][42]
ILC3s interact with the enteric nervous system to maintain intestinal homeostasis, as in response to bacteria, glial cells in the lamina propria secrete neurotrophic factors, which through the neuroregulatory receptor RET, induce IL-22 production by ILC3s.[43] Dendritic cells can also produce IL-23 during pathogen induced stress, also activating ILC3s allowing production of IL-22. One of the mechanisms by which IL-22 regulates microbiota present in the gut is through the glycosylation patterns of epithelial cells.[44] IL-22, and lymphotoxin expression by ILC3s controls expression of fucosyltransferase 2, which allows fucosylation of epithelial cells, providing a nutrient source for the luminal bacteria.[44]
AHR ligands from diet or microbiota are recognised by immune cells, regulating ILC development and NK cell functions in the intestine. In response to tryptophan metabolites, AhR signalling maintains IL-22 expression and intestinal homeostasis.[6] Retinoic acid, produced by dendritic cells, promotes the expression of gut homing receptors on ILC1s, and ILC3s, and enhances ILC3 function, by upregulating RORγt, and IL-22.[6] There is also crosstalk between macrophages and ILC3s, via RORγt driven GM-CSF production, that is dependent on microbial signalling, and the production of IL-1β by macrophages.[39] A deficiency in dietary vitamin A results in abnormally small numbers of ILC3s, and therefore a reduction of IL-22 production, and higher susceptibility to infection. Conversely, retinoic acid suppresses ILC2 proliferation by down regulating IL-7Ra, and deprivation of vitamin A has been shown to enhance ILC2- mediated resistance to helminth infection in mice.[39] ILC3s therefore form a network of interactions to maintain intestinal homeostasis, between the microbiome, intestinal epithelium, neuro-glial cells, and other immune cells.
LTi cells are present in Peyer’s Patches, and lymphoid follicles, interacting with B cells facilitating IgA production, which promotes host commensalism with the local microbiota.[45] ILC1s, and NK cells, produce IFN-γ to combat intracellular pathogens. Upon infection of C. dificile, ILC1s and ILC3s cooperate to combat the infection.[46] ILC2s induce goblet cell differentiation, and mucus production in the intestine to protect from tissue damage upon parasitic infection.
Tumor microenvironment
Different groups of Innate lymphoid cells have ability to influence tumorigenesis in several ways.[47][48]
Group 1 ILCs are the population of ILCs with the most significant anti-tumorigenic potential, with NK cells possessing the ability to recognise missing MHC Class I on the surface of tumor cells.[49] In this way, they act in a complementary manner with the cytotoxic T cells that recognize and kill tumor cells which present a foreign antigen on MHC class I.[50][51] NK cells express a number of cell surface activating NK cell receptors with specificity for stress induced ligands overexpressed on tumor cells. See the Natural killer cell page for further information on NK cells in tumor surveillance.
ILC1s influence the tumor microenvironment by the production of the cytokines IFN-γ and TNF-α, which at the beginning of immune response polarize other immune cells, such as M1 macrophages, dendritic cells, and cytotoxic T cellss to the site, creating an inflammatory environment.[52] If successful, the recruitment of these cells will kill the tumorigenic cells, however in some cases, IFN-γ and TNF-α can play a role in the induction of immunosuppressive immune cells, such as MDSCs, and therefore anti-inflammatory cytokines, allowing an immune environment the tumor cells can escape from.[53][54][48]
The role of ILC2s and ILC3s in tumor surveillance is dependent on the microenvironment encountered in their resident tissues.
ILC2s produce cytokines that promote an anti-inflammatory immune response e.g. IL-13, IL-4, Amphiregulin, favouring tumor growth.[55] However, in some settings ILC2s can produce IL-5 promoting a cytotoxic response from eosinophils and therefore an anti-tumor response.[56][57]
ILC3s can also be involved in pro or anti-tumorigenic environments. The production of IL-17 can support the growth of tumors and metastasis since it induces blood vessel permeability, however, the upregulation of MHC Class II on their surface can prime CD4+ T cells, having an anti-tumorigenic effect.[58] In addition, ILC3s have been reported to promote the formation of tertiary lymphoid structures in lung cancer, playing a protective role.[59]
Liver and metabolism
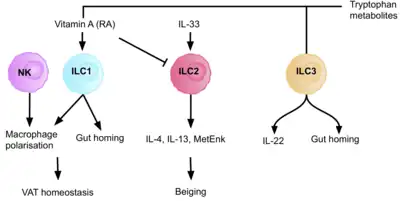
All ILC subsets are present in the liver and regulate the immune response to protect the tissue from viral and bacterial infection.[60] ILC1s are the dominant ILC subset present in the liver. Their production of IFN–γ promotes the survival of hepatocytes.[61] The production of IFN-γ by ILC1s is dependent on the expression of the NK cell receptor CD226.[61] IL-12-driven IFN-γ production by ILC1s is accelerated by extracellular ATP, and IFN-γ upregulates the prosurvival molecules Bcl-2, and Bcl-xL, in hepatocytes.[61]
NK cells play a role in the immune response against viral hepatitis B and C, limiting liver fibrosis, and liver cancer. They eliminate hepatic cells in fibrotic liver via TRAIL and/or NKG2D.
ILCs play an important role in maintaining dietary stress, and metabolic homeostasis. The production of tryptophan metabolites causes the AhR transcription factor to induce IL-22 expression, maintaining the number of ILC3s present, and therefore intestinal homeostasis.[6] The vitamin A metabolite, retinoic acid, also upregulates the expression of IL-22, and therefore, the absence of the AhR signalling pathway, and of retinoic acid, results in reduced immunity to bacterial infections, such as gastrointestinal Citrobacter rodentium infection.[6] Retinoic acid also enhances the expression of gut- homing markers on ILC1s, and ILC3s. Dietary nutrient availability therefore modifies the ILC immune response to infection and inflammation, highlighting the importance of a balanced and healthy diet.
ILC2s support a type- 2 immune environment in the adipose tissue, via the production of IL-5, IL-4 and IL-13. This regulates adiposity, insulin resistance, and caloric expenditure.[6] Dysregulation of this causes persistent type 1 inflammation, leading to obesity. ILC2s promote the beiging of adipocytes, and therefore increased energy expenditure. Therefore, decreased responses of ILC2s in the tissue are a characteristic of obesity, as this interrupts their crucial role in energy homeostasis, resulting in reduced energy expenditure and increased adiposity.[62] In addition to ILC2s, ILC1s contribute to the homeostasis of adipose tissue macrophages in both lean and obese conditions, making up 5-10% of the resident lymphocyte population, in human lean adipose depots.[10] A high fat diet increases ILC1 number, and activation of adipose tissue, increasing IFN-γ and TNF-α levels. ILC1s produce the macrophage chemoattractant CCL2, and therefore ILC1- macrophage signalling is a key regulator of adipose tissue.[63] This pathway could be a potential target for treating patients with liver disease.
Respiratory infection
ILC2s promote epithelial and goblet cell proliferation, and therefore mucus production in the respiratory tract. These functions contribute to the restoration and maintenance of epithelial integrity. ILC2s provide a defence against helminth infections in the lung, via the production of AhR, IL-9, and IL-13.[64] It is believed that these ILC2s originate in the intestine and migrate into the lung to fight the helminth infection.[65]
ILC1s and NK cells secrete IFN-γ in response to viral infection in the lungs, including rhinovirus, and respiratory syncytial virus (RSV).[3]
ILC3s are also implicated in lung infections, through the secretion of IL-17, and IL-22, for example in S. pneumoniae infection. Further studies are required to decipher the role of ILCs in human respiratory infections.[66]
Skin repair
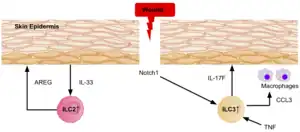
Evidence shows ILC3s and ILC2s are recruited to the wounded dermis in both mice and humans, via epidermal Notch1 signalling.[39] The ILC3s secrete IL-17F, which plays a role in the immune, and epithelial cellular responses during wound healing, by recruiting macrophages to the site. The expression of TNF also plays a role in wound healing as it directs localization of ILC3s to the damaged skin epidermis.[39] In response to the release of IL-33 by the epidermis, ILC2s secrete high levels amphiregulin, a critical epidermal growth factor, therefore contributing to cutaneous wound healing.[39]
Oral mucosa
The oral mucosa is colonized with commensals and is exposed to dietary antigens and pathogens. The ILCs in the oral mucosa help maintain the barrier and protect against infections. ILC3s and intraepithelial ILC1s were initially identified in tonsils and found in human gingivae. Approximately 10–15% of lymphocytes were identified as ILCs, most of them producing IFN-γ ILC1s. ILC3s in the oropharyngeal protect against the infection of Candida albicans producing IL-17A and IL-17F induced by IL-23. Mice lacking ILC3s due to the deletion of RORγt or depletion suffered severe infections by Candida albicans.[67]
Airways
It has been shown that the ILCs can secrete neurotransmitters and neuropeptides in the lungs. ILC2s interact with neurons in the respiratory tract by the proximity to nerve fibers, and lung resident IL-5-producing ILC2s are found in collagen-rich regions close to the confluence of medium-sized blood vessels and airways. In addition, IL-5-producing ILC2s are found in pulmonary neuroendocrine cells in the airway branch junctions at which particles entering the airways become concentrated. The localization of ILC2 in the airways suggests that the residency of ILC2 is defined by microenvironments in different zones of the tissue.[68]
Circadian circuits
The circadian clock and ILC interactions have been demonstrated by studying the regulation of the master gene clock Arntl. Its deletion resulted in the dysregulation of ILC3 caused by epigenetic changes, driving the expression of IL-22 and contributing to the alteration of the microbiome, epithelial cells, and a disrupted uptake of lipids in the intestine. On the other hand, the deletion of Nr1d1, a protein implicated in regulating circadian metabolic responses, resulted in the reduction of NCR+ ILC3 and the increase of IL-17 production, while did not affect the LTi-like ILC3.[69]
Pathology
Asthma
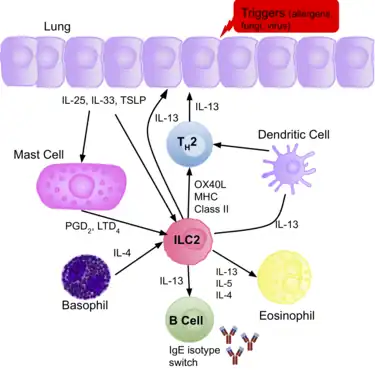
ILC2s have been confirmed to play a pathogenic role during lung inflammation. Epithelial cells in the lung express the cytokines IL-33 and IL-25, or TSLP, in response to various allergens, fungi, and viruses. These cytokines activate ILC2s, and therefore, an increased number of ILC2s, and type-2 cytokines (IL-4/5/13) are present in patients with allergic asthma.[3] They secrete IL-13, initiating allergic lung inflammation, and additionally promote Th2 differentiation, increasing the production of IL-13, and therefore amplifying the allergic response.[70]
The production of IL-5 by ILC2s in the lung leads to eosinophil recruitment, and other cell populations are known to interact and shape the presence of lung ILC2s in airway inflammation in asthmatic patients. In addition, they also promote proliferation of B cells. It is believed the increase in ILC2s present correlates with the severity of the disease, and evidence confirms some ‘allergen- experienced’ ILC2s persist after the resolution of the initial inflammation, portraying similarities to memory T cells. The presence of the ‘allergen- experienced’ ILC2s may be the reason asthmatic patients are often sensitised to various allergens.[39]
This allergic immune response appears to be independent of T and B cells, with evidence confirming that allergic responses that resembling asthma-like symptoms can be induced in mice that lack T and B cells, using IL-33.[71][72]
How other ILCs impact asthma is less clear, however studies show correlation between the number of IL-17 producing ILC3s, and the severity of the disease. It has been shown in mice that NK cells and ILC1s inhibit ILC2 expansion due to the production of IFN-γ, and therefore may help control the disease. Further research in human patients is required to determine how the balance between the different subsets impacts asthma.[73]
Autoimmune disease
NK cells express many cell-surface receptors that can be activating, inhibitory, adhesion, cytokine, or chemotactic. The integration of information collected through these numerous inputs allows NK cells to maintain self-tolerance and recognize self-cell stress signals.[74] If the nuanced, dynamic regulation of NK cell activation becomes unbalanced in favor of attacking self cells, autoimmune disease pathology. NK cell dysregulation has been implicated in a number of autoimmune disorders including multiple sclerosis, systemic lupus erythematosus, and type I diabetes mellitus.[75]
Evidence suggests that targeting ILCs may be beneficial in the design of therapeutics for autoimmune disorders. As ILCs and T cells have many redundant functions, targeting and neutralizing their effector cytokines might be a better option. Alternatively, targeting their upstream activating mediators (IL-23, IL-1B, or IL-6), or their survival factors (IL-7) could be used as an approach to treat inflammatory diseases.[21]
Allergic rhinitis
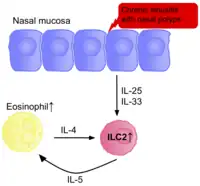
The frequency of ILC2s has also been found to be elevated in other tissues with allergic symptoms, such as the nasal polyps of patients with chronic rhinosinusitis, and in patients with aspirin exacerbated respiratory disease.[3] The concentration of ILC2s positively correlates with severity of the diseases.
ILC2s are activated due to presence of TSLP and IL-4, produced by epithelial cells and eosinophils respectively. They then produce IL-4, IL-5, and IL-13, further activating eosinophils, in a positive feedback loop, promoting inflammation. Disrupting this loop could be a potential therapy for rhinitis. NK cells appear to play a beneficial role, with fewer present in those with allergic rhinitis.[76]
Inflammatory bowel disease (IBD), and intestinal cancer
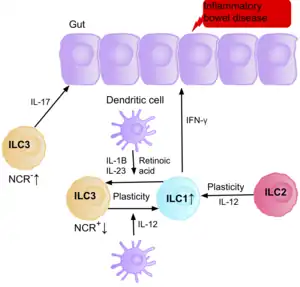
Research suggests IL-17 producing NCR- ILC3s contribute to the pathophysiology of IBD due to their increased abundance in the intestine of patients with Crohn’s disease.[39] In addition, the number of ILC1s in the intestinal mucosa of patients with Crohn’s disease is increased from approximately 10% to 40% of the total ILCs present.[39] The increase in ILCs present correlates with the severity of the disease. Evidence suggests that the plasticity between ILC3s and ILC1s in the intestine is an important factor of Crohn’s disease, with ILC3s differentiating into ILC1s when exposed to IL-12 produced by dendritic cells.[39] However, IL-23, IL-1B and retinoic acid present in the intestine can drive the differentiation of ILC1s back to ILC3s.[39] Evidence also suggests the ability of ILC2s to acquire the pro-inflammatory phenotype, with ILC2s producing IFN-γ present in the intestine of patients with Crohn’s disease, in response to certain environmental factors such as cytokines.[39]
Patients with IBD have an increased risk of getting intestinal cancer due to chronic inflammation, when the ILC3s acquire the ILC1 pro-inflammatory phenotype during chronic inflammation. Since ILCs accumulate in the intestine of IBD patients, it is believed they may have a pro-tumorigenic role. Supporting this, studies show an increase in the amount of effector cytokines IL-23, IL-17, and IL-22, in the tumor microenvironment of intestinal cancer.[77][78][79]
NK cells secrete IFN-γ, which has anti-tumorigenic effects. Multiple studies show a decreased frequency of NK cells and IFN-γ present in the intestine or peripheral blood of patients with intestinal cancer.[80][81] Further studies are required to address their exact role in the intestinal cancer environment.
Liver cancer and obesity
Hepatic ILC1s contribute to pathogenesis of chronic hepatitis B due to the production of IFN-γ, and TNF-α. Disturbance of the epithelium lining the hepatic bile ducts is frequently observed in response to chronic liver inflammation, and increased proliferation of these ducts is associated with liver cancer.[60] Evidence suggests that the enhanced proliferation is triggered by IL-13, which is produced by IL-33 induced production of ILC2 cells. ILC2s have also been shown to enhance the progression of liver fibrosis, in turn promoting the development of liver cancer.[60]
The availability of specific dietary nutrients can affect ILC immune homeostasis by altering the energy stored in the adipose tissue. Adipose tissue maintains metabolism homeostasis and is now considered a fully immunocompetent organ. Malnutrition and gluttony can dysregulate ILC responses via changes in dietary nutrients, having direct effects on the energy stored in the adipose tissue.[10] Obesity is associated with changes of gastrointestinal flora, increased afflux of free fatty acids from adipose tissue into the liver and increased gut permeability.[10] The close anatomical proximity of the gastrointestinal tract and the liver means transportation of bacterial metabolites through the portal vein triggers inflammation, acting on innate immune cells, including ILC1s, therefore playing an important role in the activation of an inflammatory state in the liver. Therefore, inflammation associated with obesity can influence the progression of liver disease, due to the development of insulin resistance and metabolic dysregulation.[10] ILC1s as a key regulatory of adipose tissue inflammation, are therefore a potential therapeutic target for treating people with liver disease or metabolic syndrome.
ILC2s have also been identified in human and mouse white adipose tissue, contributing to the development of obesity. Upon dysregulation of homeostasis in the adipose tissue, the decreased responses of ILC2s are a characteristic of obesity, as this interrupts their crucial role in energy homeostasis, resulting in reduced energy expenditure, and increased adiposity.[62]
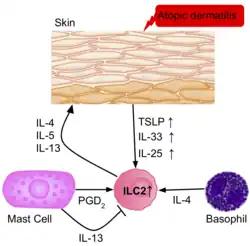
Skin inflammation
The frequency of ILC2s is higher in the inflamed skin of patients with atopic dermatitis than in healthy patients.[39] The ILC2s from the skin of the patients had upregulation of the IL-25, IL-33, TSLP and PGD2 receptors, suggesting their role in the activation of ILC2s. Basophils and mast cells are also present in these skin lesions, producing IL-4, and PGD2, further activating ILC2s.
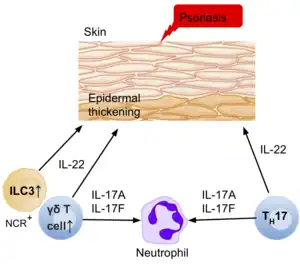
Psoriasis, another inflammatory skin disease, causes epidermal thickening, forming plaques which are mainly populated with T cells and dendritic cells. The T cells portray a type 1 immune response; however, the thickening and inflammation of the epidermis is thought to be caused by the production of IL-22, IL-17A, and IL-17F by other T cells such as Th17 or γδ T cells.[39] However, more recent data suggests that ILC3s in fact produce a large number of these cytokines, with an increase in the number of ILC3s in the peripheral blood of patients with psoriasis.[39]
Arthritis
The ILCs have been studied in mucosal barriers and their interplay with adaptative immunity, thus implicating them with autoimmune diseases. In arthritis characterized by autoantibodies presence, the dysregulated crosstalk between Tfh and B cells has been implicated in generating those antibodies. Interestingly, it has been suggested that Th17 and Tfh inflammatory responses are generated in the gastrointestinal tract and that microbiota can increase this response. Thus the development of ILCs implicated in regulating the immune response against the microbiota in the intestine has been associated with arthritis. In case of ILC2 has an important role in regulating inflammatory responses by producing IL-4, IL-9, and IL-13.[82]
Multiple sclerosis
In the case of ILC3 in multiple sclerosis, these cells have been implicated with tertiary lymphoid aggregates in the brain of patients with progressive disease. In addition, the increase of LTi-like ILC3 correlated with the autoantibodies in the brain fluid.[82]
Plasticity
Our classification of ILCs into subsets provides a simplified framework, however, despite the above classification system, several studies suggest their development and phenotypic maintenance is much more complex, with a high level of plasticity between the subsets. Studies have confirmed the ability of some ILC subsets to convert into a different subset in the presence of specific cytokines.[13][47] This is also a common feature in T cells, and it is believed this plasticity is critical to allow our immune system to fine tune responses to so many different pathogens.[13] ILC plasticity requires cytokine receptors, their transcription factors, and access of defined chromatin regions to the transcription factors, however, it still remains unclear where these cytokines are produced and where the differentiation occurs in Vivo.[6]
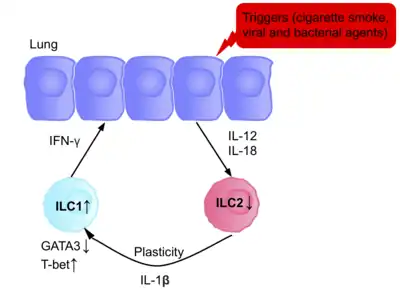
The ILCs present in patients with chronic obstructive pulmonary disease (COPD) are a prototypical example of ILC plasticity. Studies in both humans and mice have shown lung resident ILC2s acquire an ILC1 phenotype during COPD, increasing IFN-γ secretion, and therefore inflammation.[83] Various triggers, including cigarette smoke, cause secretion of IL-12 and IL-18, causing the differentiation ILC2s into ILC1s. GATA3 is down-regulated, and T-bet expression is up-regulated.[83] Patients therefore have a higher blood ILC1:ILC2 ratio, with the abundance of ILC1s present correlating with the severity of the disease.[83]
The ability of ILC3s to convert into ILC1-like cells has been shown in vitro, and in vivo.[84][85][47] When ILC3s are cultured with IL-2 and IL-15, it causes the up-regulation of T-bet, and the IL-12 receptor (IL-12R) β2, allowing conversion of ILC3s to ILC1s. In addition, studies suggest IL-23 can promote the conversion of ILC1s into ILC3s.[85]
There is increasing evidence indicating that ILC2s also have a certain degree of plasticity, with studies confirming their ability to convert into ILC1s and ILC3s upon exposure to specific environmental stimuli such as cytokines, or notch ligands.[86][47]
The signaling induced by the cytokines governs the plasticity between ILC3 and ILC1, inducing the expression of T-bet. In patients with Crohn’s disease, the increase of ILC1 at the expense of ILC3 possibly by the production of IL-2 from T regulatory cell, leading to a pathogenic state and inflammatory events. Although the plasticity is reversible, during the differentiation of NKp46+ ILC3s to ILC1, the modulation of the expression of T-bet depends on IL-23, IL-2, and IL-1b and is improved by retinoic acid. Therefore, ILC3 to ILC1 plasticity depends on dendritic cells that produce these cytokines. Although the interconversion of ILC1 and ILC3 is modulated by the differential expression of RORγt and T-bet, different questions remain that need to be explained to understand the inflammation caused by these cells.[87]
In the case of ILC2, Gata3 can be downregulated due to the exposure of infectious agents such as the influenza virus, respiratory syncytial virus, and Staphylococcus aureus, increasing the expression of IL12Rb2, IL-18Ra, and T-bet. The differentiation of ILC2 to ILC1 can also be reversible, although the mechanism is not understood yet.[87]
In certain environments, such as inflammation, chronic disease, or tumor microenvironments, activated NK cells can start to express CD49a, and CXCR6, common ILC1 markers, strengthening their plastic properties.[88][89]
Determining the extent of ILC plasticity during disease could be useful to allow us to prevent or enhance their conversion into other subsets that may be contributing to the pathogenicity.[47][90]
Innate or adaptive
Historically, the distinction between the innate and adaptive immune system focused on the innate system’s nonspecific nature and lack of memory.[91] As information has emerged about the functions of NK cells and other ILCs as effectors and orchestrators of the adaptive immune response, this distinction has become less clear. Some researchers suggest that the definition should focus more on the germline-coding of receptors in the innate immune system versus the rearranged receptors of the adaptive immune system.[74]
See also
References
- Spits H, Cupedo T (2012). "Innate lymphoid cells: emerging insights in development, lineage relationships, and function". Annual Review of Immunology. 30: 647–75. doi:10.1146/annurev-immunol-020711-075053. PMID 22224763.
- Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. (February 2013). "Innate lymphoid cells--a proposal for uniform nomenclature". Nature Reviews. Immunology. 13 (2): 145–9. doi:10.1038/nri3365. PMID 23348417. S2CID 2228459.
- Panda SK, Colonna M (2019). "Innate Lymphoid Cells in Mucosal Immunity". Frontiers in Immunology. 10: 861. doi:10.3389/fimmu.2019.00861. PMC 6515929. PMID 31134050.
- Walker JA, Barlow JL, McKenzie AN (February 2013). "Innate lymphoid cells--how did we miss them?". Nature Reviews. Immunology. 13 (2): 75–87. doi:10.1038/nri3349. PMID 23292121. S2CID 14580303.
- Klose CS, Kiss EA, Schwierzeck V, Ebert K, Hoyler T, d'Hargues Y, et al. (February 2013). "A T-bet gradient controls the fate and function of CCR6-RORγt+ innate lymphoid cells". Nature. 494 (7436): 261–5. Bibcode:2013Natur.494..261K. doi:10.1038/nature11813. PMID 23334414. S2CID 4390857.
- Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. (August 2018). "Innate Lymphoid Cells: 10 Years On". Cell. 174 (5): 1054–1066. doi:10.1016/j.cell.2018.07.017. PMID 30142344.
- Jowett GM, Norman MD, Yu TT, Rosell Arévalo P, Hoogland D, Lust ST, et al. (February 2021). "ILC1 drive intestinal epithelial and matrix remodelling". Nature Materials. 20 (2): 250–259. doi:10.1038/s41563-020-0783-8. PMC 7611574. PMID 32895507. S2CID 221521946.
- Daussy C, Faure F, Mayol K, Viel S, Gasteiger G, Charrier E, et al. (March 2014). "T-bet and Eomes instruct the development of two distinct natural killer cell lineages in the liver and in the bone marrow". The Journal of Experimental Medicine. 211 (3): 563–77. doi:10.1084/jem.20131560. PMC 3949572. PMID 24516120.
- Simonetta F, Pradier A, Roosnek E (2016). "T-bet and Eomesodermin in NK Cell Development, Maturation, and Function". Frontiers in Immunology. 7: 241. doi:10.3389/fimmu.2016.00241. PMC 4913100. PMID 27379101.
- Luci C, Vieira E, Perchet T, Gual P, Golub R (2019). "Natural Killer Cells and Type 1 Innate Lymphoid Cells Are New Actors in Non-alcoholic Fatty Liver Disease". Frontiers in Immunology. 10: 1192. doi:10.3389/fimmu.2019.01192. PMC 6546848. PMID 31191550.
- Weizman OE, Adams NM, Schuster IS, Krishna C, Pritykin Y, Lau C, et al. (November 2017). "ILC1 Confer Early Host Protection at Initial Sites of Viral Infection". Cell. 171 (4): 795–808.e12. doi:10.1016/j.cell.2017.09.052. PMC 5687850. PMID 29056343.
- Cortez VS, Fuchs A, Cella M, Gilfillan S, Colonna M (May 2014). "Cutting edge: Salivary gland NK cells develop independently of Nfil3 in steady-state". Journal of Immunology. 192 (10): 4487–91. doi:10.4049/jimmunol.1303469. PMID 24740507.
- Colonna M (June 2018). "Innate Lymphoid Cells: Diversity, Plasticity, and Unique Functions in Immunity". Immunity. 48 (6): 1104–1117. doi:10.1016/j.immuni.2018.05.013. PMC 6344351. PMID 29924976.
- Kim BS, Siracusa MC, Saenz SA, Noti M, Monticelli LA, Sonnenberg GF, et al. (January 2013). "TSLP elicits IL-33-independent innate lymphoid cell responses to promote skin inflammation". Science Translational Medicine. 5 (170): 170ra16. doi:10.1126/scitranslmed.3005374. PMC 3637661. PMID 23363980.
- Roediger B, Kyle R, Yip KH, Sumaria N, Guy TV, Kim BS, et al. (June 2013). "Cutaneous immunosurveillance and regulation of inflammation by group 2 innate lymphoid cells". Nature Immunology. 14 (6): 564–73. doi:10.1038/ni.2584. PMC 4282745. PMID 23603794.
- Neill DR, Wong SH, Bellosi A, Flynn RJ, Daly M, Langford TK, et al. (April 2010). "Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity". Nature. 464 (7293): 1367–70. Bibcode:2010Natur.464.1367N. doi:10.1038/nature08900. PMC 2862165. PMID 20200518.
- Mjösberg J, Bernink J, Golebski K, Karrich JJ, Peters CP, Blom B, et al. (October 2012). "The transcription factor GATA3 is essential for the function of human type 2 innate lymphoid cells". Immunity. 37 (4): 649–59. doi:10.1016/j.immuni.2012.08.015. PMID 23063330.
- Juelke K, Romagnani C (February 2016). "Differentiation of human innate lymphoid cells (ILCs)". Current Opinion in Immunology. 38: 75–85. doi:10.1016/j.coi.2015.11.005. PMID 26707651.
- Buonocore S, Ahern PP, Uhlig HH, Ivanov II, Littman DR, Maloy KJ, Powrie F (April 2010). "Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology". Nature. 464 (7293): 1371–5. Bibcode:2010Natur.464.1371B. doi:10.1038/nature08949. PMC 3796764. PMID 20393462.
- Gaffen SL, Jain R, Garg AV, Cua DJ (September 2014). "The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing". Nature Reviews. Immunology. 14 (9): 585–600. doi:10.1038/nri3707. PMC 4281037. PMID 25145755.
- Pantazi E, Powell N (2019). "Group 3 ILCs: Peacekeepers or Troublemakers? What's Your Gut Telling You?!". Frontiers in Immunology. 10: 676. doi:10.3389/fimmu.2019.00676. PMC 6460375. PMID 31024537.
- Cupedo T, Crellin NK, Papazian N, Rombouts EJ, Weijer K, Grogan JL, et al. (January 2009). "Human fetal lymphoid tissue-inducer cells are interleukin 17-producing precursors to RORC+ CD127+ natural killer-like cells". Nature Immunology. 10 (1): 66–74. doi:10.1038/ni.1668. PMID 19029905. S2CID 22864899.
- Takatori H, Kanno Y, Watford WT, Tato CM, Weiss G, Ivanov II, et al. (January 2009). "Lymphoid tissue inducer-like cells are an innate source of IL-17 and IL-22". The Journal of Experimental Medicine. 206 (1): 35–41. doi:10.1084/jem.20072713. PMC 2626689. PMID 19114665.
- Withers DR (May 2011). "Lymphoid tissue inducer cells". Current Biology. 21 (10): R381-2. doi:10.1016/j.cub.2011.03.022. PMID 21601793.
- Mebius RE, Rennert P, Weissman IL (October 1997). "Developing lymph nodes collect CD4+CD3- LTbeta+ cells that can differentiate to APC, NK cells, and follicular cells but not T or B cells". Immunity. 7 (4): 493–504. doi:10.1016/S1074-7613(00)80371-4. PMID 9354470.
- Strober W (November 2010). "The LTi cell, an immunologic chameleon". Immunity. 33 (5): 650–2. doi:10.1016/j.immuni.2010.11.016. PMC 3426921. PMID 21094460.
- Eberl G, Colonna M, Di Santo JP, McKenzie AN (May 2015). "Innate lymphoid cells. Innate lymphoid cells: a new paradigm in immunology". Science. 348 (6237): aaa6566. doi:10.1126/science.aaa6566. PMC 5658207. PMID 25999512.
- Klose CS, Flach M, Möhle L, Rogell L, Hoyler T, Ebert K, et al. (April 2014). "Differentiation of type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages". Cell. 157 (2): 340–356. doi:10.1016/j.cell.2014.03.030. PMID 24725403.
- Xu W, Domingues RG, Fonseca-Pereira D, Ferreira M, Ribeiro H, Lopez-Lastra S, et al. (March 2015). "NFIL3 orchestrates the emergence of common helper innate lymphoid cell precursors". Cell Reports. 10 (12): 2043–54. doi:10.1016/j.celrep.2015.02.057. PMID 25801035.
- Bando JK, Liang HE, Locksley RM (February 2015). "Identification and distribution of developing innate lymphoid cells in the fetal mouse intestine". Nature Immunology. 16 (2): 153–60. doi:10.1038/ni.3057. PMC 4297560. PMID 25501629.
- Lee JS, Cella M, McDonald KG, Garlanda C, Kennedy GD, Nukaya M, et al. (November 2011). "AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of Notch". Nature Immunology. 13 (2): 144–51. doi:10.1038/ni.2187. PMC 3468413. PMID 22101730.
- Kotas ME, Locksley RM (June 2018). "Why Innate Lymphoid Cells?". Immunity. 48 (6): 1081–1090. doi:10.1016/j.immuni.2018.06.002. PMC 6145487. PMID 29924974.
- Löser S, Smith KA, Maizels RM (2019). "Innate Lymphoid Cells in Helminth Infections-Obligatory or Accessory?". Frontiers in Immunology. 10: 620. doi:10.3389/fimmu.2019.00620. PMC 6467944. PMID 31024526.
- Palm NW, Rosenstein RK, Medzhitov R (April 2012). "Allergic host defences". Nature. 484 (7395): 465–72. Bibcode:2012Natur.484..465P. doi:10.1038/nature11047. PMC 3596087. PMID 22538607.
- Dahlgren MW, Jones SW, Cautivo KM, Dubinin A, Ortiz-Carpena JF, Farhat S, et al. (March 2019). "Adventitial Stromal Cells Define Group 2 Innate Lymphoid Cell Tissue Niches". Immunity. 50 (3): 707–722.e6. doi:10.1016/j.immuni.2019.02.002. PMC 6553479. PMID 30824323.
- Sui P, Wiesner DL, Xu J, Zhang Y, Lee J, Van Dyken S, et al. (June 2018). "Pulmonary neuroendocrine cells amplify allergic asthma responses". Science. 360 (6393): eaan8546. doi:10.1126/science.aan8546. PMC 6387886. PMID 29599193.
- Bernink JH, Peters CP, Munneke M, te Velde AA, Meijer SL, Weijer K, et al. (March 2013). "Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues". Nature Immunology. 14 (3): 221–9. doi:10.1038/ni.2534. PMID 23334791. S2CID 8614680.
- Willinger T (2019). "Metabolic Control of Innate Lymphoid Cell Migration". Frontiers in Immunology. 10: 2010. doi:10.3389/fimmu.2019.02010. PMC 6713999. PMID 31507605.
- Ebbo M, Crinier A, Vély F, Vivier E (November 2017). "Innate lymphoid cells: major players in inflammatory diseases". Nature Reviews. Immunology. 17 (11): 665–678. doi:10.1038/nri.2017.86. PMID 28804130. S2CID 2651328.
- Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, et al. (March 2008). "Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens". Nature Medicine. 14 (3): 282–9. doi:10.1038/nm1720. PMID 18264109. S2CID 15742387.
- Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. (September 2006). "The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells". Cell. 126 (6): 1121–33. doi:10.1016/j.cell.2006.07.035. PMID 16990136. S2CID 9034013.
- Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, et al. (September 2007). "IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways". Nature Immunology. 8 (9): 967–74. doi:10.1038/ni1488. PMID 17581537. S2CID 21177884.
- Ibiza S, García-Cassani B, Ribeiro H, Carvalho T, Almeida L, Marques R, et al. (July 2016). "Glial-cell-derived neuroregulators control type 3 innate lymphoid cells and gut defence". Nature. 535 (7612): 440–443. Bibcode:2016Natur.535..440I. doi:10.1038/nature18644. PMC 4962913. PMID 27409807.
- Goto Y, Obata T, Kunisawa J, Sato S, Ivanov II, Lamichhane A, et al. (September 2014). "Innate lymphoid cells regulate intestinal epithelial cell glycosylation". Science. 345 (6202): 1254009. doi:10.1126/science.1254009. PMC 4774895. PMID 25214634.
- Macpherson AJ, Yilmaz B, Limenitakis JP, Ganal-Vonarburg SC (April 2018). "IgA Function in Relation to the Intestinal Microbiota". Annual Review of Immunology. 36 (1): 359–381. doi:10.1146/annurev-immunol-042617-053238. PMID 29400985.
- Abt MC, Lewis BB, Caballero S, Xiong H, Carter RA, Sušac B, et al. (July 2015). "Innate Immune Defenses Mediated by Two ILC Subsets Are Critical for Protection against Acute Clostridium difficile Infection". Cell Host & Microbe. 18 (1): 27–37. doi:10.1016/j.chom.2015.06.011. PMC 4537644. PMID 26159718.
- Wagner M, Moro K, Koyasu S (May 2017). "Plastic Heterogeneity of Innate Lymphoid Cells in Cancer". Trends in Cancer. 3 (5): 326–335. doi:10.1016/j.trecan.2017.03.008. PMID 28718410.
- Wagner M, Koyasu S (May 2019). "Cancer Immunoediting by Innate Lymphoid Cells". Trends in Immunology. 40 (5): 415–430. doi:10.1016/j.it.2019.03.004. PMID 30992189. S2CID 119093972.
- Dadi S, Chhangawala S, Whitlock BM, Franklin RA, Luo CT, Oh SA, et al. (January 2016). "Cancer Immunosurveillance by Tissue-Resident Innate Lymphoid Cells and Innate-like T Cells". Cell. 164 (3): 365–77. doi:10.1016/j.cell.2016.01.002. PMC 4733424. PMID 26806130.
- Cerwenka A, Lanier LL (October 2001). "Natural killer cells, viruses and cancer". Nature Reviews. Immunology. 1 (1): 41–9. doi:10.1038/35095564. PMID 11905813. S2CID 205021117.
- Smyth MJ, Godfrey DI, Trapani JA (April 2001). "A fresh look at tumor immunosurveillance and immunotherapy". Nature Immunology. 2 (4): 293–9. doi:10.1038/86297. PMID 11276199. S2CID 24779449.
- Fuchs A, Vermi W, Lee JS, Lonardi S, Gilfillan S, Newberry RD, et al. (April 2013). "Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12- and IL-15-responsive IFN-γ-producing cells". Immunity. 38 (4): 769–81. doi:10.1016/j.immuni.2013.02.010. PMC 3634355. PMID 23453631.
- Lechner MG, Liebertz DJ, Epstein AL (August 2010). "Characterization of cytokine-induced myeloid-derived suppressor cells from normal human peripheral blood mononuclear cells". Journal of Immunology. 185 (4): 2273–84. doi:10.4049/jimmunol.1000901. PMC 2923483. PMID 20644162.
- Heeren, A. Marijne, et al. "High and interrelated rates of PD-L1+ CD14+ antigen-presenting cells and regulatory T cells mark the microenvironment of metastatic lymph nodes from patients with cervical cancer." Cancer immunology research (2014): canimm-0149.
- Zhu J (September 2015). "T helper 2 (Th2) cell differentiation, type 2 innate lymphoid cell (ILC2) development and regulation of interleukin-4 (IL-4) and IL-13 production". Cytokine. 75 (1): 14–24. doi:10.1016/j.cyto.2015.05.010. PMC 4532589. PMID 26044597.
- Ikutani M, Yanagibashi T, Ogasawara M, Tsuneyama K, Yamamoto S, Hattori Y, et al. (January 2012). "Identification of innate IL-5-producing cells and their role in lung eosinophil regulation and antitumor immunity". Journal of Immunology. 188 (2): 703–13. doi:10.4049/jimmunol.1101270. PMID 22174445.
- Wagner M, Ealey KN, Tetsu H, Kiniwa T, Motomura Y, Moro K, Koyasu S (February 2020). "Tumor-Derived Lactic Acid Contributes to the Paucity of Intratumoral ILC2s". Cell Reports. 30 (8): 2743–2757.e5. doi:10.1016/j.celrep.2020.01.103. PMID 32101749.
- Ducimetière L, Vermeer M, Tugues S (2019). "The Interplay Between Innate Lymphoid Cells and the Tumor Microenvironment". Frontiers in Immunology. 10: 2895. doi:10.3389/fimmu.2019.02895. PMC 6923277. PMID 31921156.
- Carrega P, Loiacono F, Di Carlo E, Scaramuccia A, Mora M, Conte R, et al. (September 2015). "NCR(+)ILC3 concentrate in human lung cancer and associate with intratumoral lymphoid structures". Nature Communications. 6 (1): 8280. Bibcode:2015NatCo...6.8280C. doi:10.1038/ncomms9280. PMID 26395069.
- Ochel A, Tiegs G, Neumann K (April 2019). "Type 2 Innate Lymphoid Cells in Liver and Gut: From Current Knowledge to Future Perspectives". International Journal of Molecular Sciences. 20 (8): 1896. doi:10.3390/ijms20081896. PMC 6514972. PMID 30999584.
- Nabekura T, Riggan L, Hildreth AD, O'Sullivan TE, Shibuya A (January 2020). "Type 1 Innate Lymphoid Cells Protect Mice from Acute Liver Injury via Interferon-γ Secretion for Upregulating Bcl-xL Expression in Hepatocytes". Immunity. 52 (1): 96–108.e9. doi:10.1016/j.immuni.2019.11.004. PMC 8108607. PMID 31810881.
- Brestoff JR, Kim BS, Saenz SA, Stine RR, Monticelli LA, Sonnenberg GF, et al. (March 2015). "Group 2 innate lymphoid cells promote beiging of white adipose tissue and limit obesity". Nature. 519 (7542): 242–6. Bibcode:2015Natur.519..242B. doi:10.1038/nature14115. PMC 4447235. PMID 25533952.
- Lee BC, Kim MS, Pae M, Yamamoto Y, Eberlé D, Shimada T, et al. (April 2016). "Adipose Natural Killer Cells Regulate Adipose Tissue Macrophages to Promote Insulin Resistance in Obesity". Cell Metabolism. 23 (4): 685–98. doi:10.1016/j.cmet.2016.03.002. PMC 4833527. PMID 27050305.
- Turner JE, Morrison PJ, Wilhelm C, Wilson M, Ahlfors H, Renauld JC, et al. (December 2013). "IL-9-mediated survival of type 2 innate lymphoid cells promotes damage control in helminth-induced lung inflammation". The Journal of Experimental Medicine. 210 (13): 2951–65. doi:10.1084/jem.20130071. PMC 3865473. PMID 24249111.
- Huang Y, Mao K, Chen X, Sun MA, Kawabe T, Li W, et al. (January 2018). "S1P-dependent interorgan trafficking of group 2 innate lymphoid cells supports host defense". Science. 359 (6371): 114–119. Bibcode:2018Sci...359..114H. doi:10.1126/science.aam5809. PMC 6956613. PMID 29302015.
- Van Maele L, Carnoy C, Cayet D, Ivanov S, Porte R, Deruy E, et al. (August 2014). "Activation of Type 3 innate lymphoid cells and interleukin 22 secretion in the lungs during Streptococcus pneumoniae infection". The Journal of Infectious Diseases. 210 (3): 493–503. doi:10.1093/infdis/jiu106. PMID 24577508.
- Panda SK, Colonna M (2019-05-07). "Innate Lymphoid Cells in Mucosal Immunity". Frontiers in Immunology. 10: 861. doi:10.3389/fimmu.2019.00861. PMC 6515929. PMID 31134050.
- Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. (August 2018). "Innate Lymphoid Cells: 10 Years On". Cell. 174 (5): 1054–1066. doi:10.1016/j.cell.2018.07.017. PMID 30142344. S2CID 52079371.
- Domingues RG, Hepworth MR (2020-02-06). "Immunoregulatory Sensory Circuits in Group 3 Innate Lymphoid Cell (ILC3) Function and Tissue Homeostasis". Frontiers in Immunology. 11: 116. doi:10.3389/fimmu.2020.00116. PMC 7015949. PMID 32117267.
- Halim TY, Steer CA, Mathä L, Gold MJ, Martinez-Gonzalez I, McNagny KM, et al. (March 2014). "Group 2 innate lymphoid cells are critical for the initiation of adaptive T helper 2 cell-mediated allergic lung inflammation". Immunity. 40 (3): 425–35. doi:10.1016/j.immuni.2014.01.011. PMC 4210641. PMID 24613091.
- Oboki K, Nakae S, Matsumoto K, Saito H (April 2011). "IL-33 and Airway Inflammation". Allergy, Asthma & Immunology Research. 3 (2): 81–8. doi:10.4168/aair.2011.3.2.81. PMC 3062800. PMID 21461246.
- Kondo H, Ichikawa Y, Imokawa G (March 1998). "Percutaneous sensitization with allergens through barrier-disrupted skin elicits a Th2-dominant cytokine response". European Journal of Immunology. 28 (3): 769–79. doi:10.1002/(SICI)1521-4141(199803)28:03<769::AID-IMMU769>3.0.CO;2-H. PMID 9541570. S2CID 21654970.
- Kim HY, Lee HJ, Chang YJ, Pichavant M, Shore SA, Fitzgerald KA, et al. (January 2014). "Interleukin-17-producing innate lymphoid cells and the NLRP3 inflammasome facilitate obesity-associated airway hyperreactivity". Nature Medicine. 20 (1): 54–61. doi:10.1038/nm.3423. PMC 3912313. PMID 24336249.
- Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, et al. (January 2011). "Innate or adaptive immunity? The example of natural killer cells". Science. 331 (6013): 44–9. Bibcode:2011Sci...331...44V. doi:10.1126/science.1198687. PMC 3089969. PMID 21212348.
- Baxter AG, Smyth MJ (February 2002). "The role of NK cells in autoimmune disease". Autoimmunity. 35 (1): 1–14. doi:10.1080/08916930290005864. PMID 11908701. S2CID 28199633.
- Scordamaglia F, Balsamo M, Scordamaglia A, Moretta A, Mingari MC, Canonica GW, et al. (February 2008). "Perturbations of natural killer cell regulatory functions in respiratory allergic diseases". The Journal of Allergy and Clinical Immunology. 121 (2): 479–85. doi:10.1016/j.jaci.2007.09.047. PMID 18061653.
- Langowski JL, Zhang X, Wu L, Mattson JD, Chen T, Smith K, et al. (July 2006). "IL-23 promotes tumour incidence and growth". Nature. 442 (7101): 461–5. Bibcode:2006Natur.442..461L. doi:10.1038/nature04808. PMID 16688182. S2CID 4431794.
- Wu S, Rhee KJ, Albesiano E, Rabizadeh S, Wu X, Yen HR, et al. (September 2009). "A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses". Nature Medicine. 15 (9): 1016–22. doi:10.1038/nm.2015. PMC 3034219. PMID 19701202.
- Grivennikov SI, Wang K, Mucida D, Stewart CA, Schnabl B, Jauch D, et al. (November 2012). "Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth". Nature. 491 (7423): 254–8. Bibcode:2012Natur.491..254G. doi:10.1038/nature11465. PMC 3601659. PMID 23034650.
- Bie Q, Zhang P, Su Z, Zheng D, Ying X, Wu Y, et al. (2014). "Polarization of ILC2s in peripheral blood might contribute to immunosuppressive microenvironment in patients with gastric cancer". Journal of Immunology Research. 2014: 923135. doi:10.1155/2014/923135. PMC 3987940. PMID 24741632.
- Lee J, Park KH, Ryu JH, Bae HJ, Choi A, Lee H, et al. (September 2017). "Natural killer cell activity for IFN-gamma production as a supportive diagnostic marker for gastric cancer". Oncotarget. 8 (41): 70431–70440. doi:10.18632/oncotarget.19712. PMC 5642566. PMID 29050291.
- Sonnenberg GF, Hepworth MR (October 2019). "Functional interactions between innate lymphoid cells and adaptive immunity". Nature Reviews. Immunology. 19 (10): 599–613. doi:10.1038/s41577-019-0194-8. PMC 6982279. PMID 31350531.
- Bal SM, Bernink JH, Nagasawa M, Groot J, Shikhagaie MM, Golebski K, et al. (June 2016). "IL-1β, IL-4 and IL-12 control the fate of group 2 innate lymphoid cells in human airway inflammation in the lungs". Nature Immunology. 17 (6): 636–45. doi:10.1038/ni.3444. PMID 27111145. S2CID 883747.
- Cella M, Otero K, Colonna M (June 2010). "Expansion of human NK-22 cells with IL-7, IL-2, and IL-1beta reveals intrinsic functional plasticity". Proceedings of the National Academy of Sciences of the United States of America. 107 (24): 10961–6. Bibcode:2010PNAS..10710961C. doi:10.1073/pnas.1005641107. PMC 2890739. PMID 20534450.
- Bernink JH, Krabbendam L, Germar K, de Jong E, Gronke K, Kofoed-Nielsen M, et al. (July 2015). "Interleukin-12 and -23 Control Plasticity of CD127(+) Group 1 and Group 3 Innate Lymphoid Cells in the Intestinal Lamina Propria". Immunity. 43 (1): 146–60. doi:10.1016/j.immuni.2015.06.019. PMID 26187413.
- Zhang K, Xu X, Pasha MA, Siebel CW, Costello A, Haczku A, et al. (March 2017). "Cutting Edge: Notch Signaling Promotes the Plasticity of Group-2 Innate Lymphoid Cells". Journal of Immunology. 198 (5): 1798–1803. doi:10.4049/jimmunol.1601421. PMC 5321819. PMID 28115527.
- Meininger I, Carrasco A, Rao A, Soini T, Kokkinou E, Mjösberg J (October 2020). "Tissue-Specific Features of Innate Lymphoid Cells". Trends in Immunology. 41 (10): 902–917. doi:10.1016/j.it.2020.08.009. PMID 32917510. S2CID 221636264.
- Gao Y, Souza-Fonseca-Guimaraes F, Bald T, Ng SS, Young A, Ngiow SF, et al. (September 2017). "Tumor immunoevasion by the conversion of effector NK cells into type 1 innate lymphoid cells". Nature Immunology. 18 (9): 1004–1015. doi:10.1038/ni.3800. PMID 28759001. S2CID 30239.
- Cortez VS, Ulland TK, Cervantes-Barragan L, Bando JK, Robinette ML, Wang Q, et al. (September 2017). "SMAD4 impedes the conversion of NK cells into ILC1-like cells by curtailing non-canonical TGF-β signaling". Nature Immunology. 18 (9): 995–1003. doi:10.1038/ni.3809. PMC 5712491. PMID 28759002.
- Bald T, Wagner M, Gao Y, Koyasu S, Smyth MJ (February 2019). "Hide and seek: Plasticity of innate lymphoid cells in cancer". Seminars in Immunology. 41: 101273. doi:10.1016/j.smim.2019.04.001. PMID 30979591. S2CID 111390262.
- Lanier LL (February 2013). "Shades of grey--the blurring view of innate and adaptive immunity" (PDF). Nature Reviews. Immunology. 13 (2): 73–4. doi:10.1038/nri3389. PMID 23469373. S2CID 27204420.