Hereditary nonpolyposis colorectal cancer
Hereditary nonpolyposis colorectal cancer (HNPCC) or Lynch syndrome is an autosomal dominant genetic condition that is associated with a high risk of colon cancer as well as other cancers including endometrial cancer (second most common), ovary, stomach, small intestine, hepatobiliary tract, upper urinary tract, brain, and skin.[2] The increased risk for these cancers is due to inherited genetic mutations that impair DNA mismatch repair. It is a type of cancer syndrome. Because patients with Lynch syndrome can have polyps, the term HNPCC has fallen out of favor.
| Hereditary nonpolyposis colorectal cancer | |
|---|---|
| Other names | Lynch syndrome[1] |
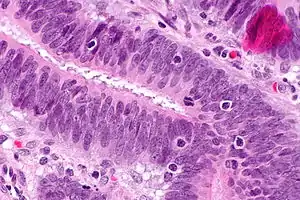 | |
| Micrograph showing tumor-infiltrating lymphocytes (in a colorectal cancer), a finding associated with MSI-H tumours, as may be seen in Lynch syndrome. H&E stain. | |
| Specialty | Oncology |
Signs and symptoms
Risk of cancer
Lifetime risk and mean age at diagnosis for Lynch syndrome associated cancers[3]
| Type of cancer | Lifetime risk (%) | Mean age at diagnosis (years) |
| Colorectal | 52-58 | 44-61 |
| Endometrial | 25-60 | 48-62 |
| Gastric | 6-13 | 56 |
| Ovarian | 4-12 | 42.5 |
In addition to the types of cancer found in the chart above, it is understood that Lynch syndrome also contributes to an increased risk of small bowel cancer, pancreatic cancer, ureter/renal pelvis cancer, biliary tract cancer, brain cancer, and sebaceous neoplasms.[3] Increased risk of prostate cancer and breast cancer has also been associated with Lynch syndrome, although this relationship is not entirely understood.[3]
Two-thirds of colon cancers occur in the proximal colon and common signs and symptoms include blood in the stool, diarrhea or constipation, and unintended weight loss.[4] The mean age of colorectal cancer diagnosis is 44 for members of families that meet the Amsterdam criteria. The average age of diagnosis of endometrial cancer is about 46 years. Among women with HNPCC who have both colon and endometrial cancer, about half present first with endometrial cancer, making endometrial cancer the most common sentinel cancer in Lynch syndrome.[5] The most common symptom of endometrial cancer is abnormal vaginal bleeding.[6] In HNPCC, the mean age of diagnosis of gastric cancer is 56 years of age with intestinal-type adenocarcinoma being the most commonly reported pathology. HNPCC-associated ovarian cancers have an average age of diagnosis of 42.5 years-old; approximately 30% are diagnosed before age 40.
Significant variation in the rate of cancer has been found depending on the mutation involved.[7][8] Up to the age of 75 years the risks of different cancers by the mutations are in the table below.
| Gene | colorectal cancer risk | endometrial cancer risk | ovarian cancer risk | upper gastrointestinal (gastric, duodenal, bile duct or pancreatic) cancer risk | urinary tract cancers risk | prostate cancer risk | brain tumor risk |
| MLH1 | 46% | 43% | 10% | 21% | 8% | 17% | 1% |
| MSH2 | 57% | 17% | 10% | 25% | 32% | 5% | n.a. |
| MSH6 | 15% | 46% | 13% | 7% | 11% | 18% | 1% |
| Gene | Ovarian cancer risk | Endometrial cancer risk |
|---|---|---|
| MLH1 | 4-24% | 25-60% |
| MSH2/EPCAM | 4-24% | 25-60% |
| MSH6 | 1-11% | 16-26% |
| PMS2 | 6% (combined risk) | 15% |
Genetics

HNPCC is inherited in an autosomal dominant fashion.[10] The hallmark of HNPCC is defective DNA mismatch repair, which causes an elevated rate of single nucleotide changes and microsatellite instability, also known as MSI-H (the H is "high"). MSI is identifiable in cancer specimens in the pathology laboratory.[11] Most cases result in changes in the lengths of dinucleotide repeats of the nucleobases cytosine and adenine (sequence: CACACACACA...).[12]
The 4 main genes involved in HNPCC normally encode for proteins that form dimers to function:
- MLH1 protein dimerizes with PMS2 protein to form MutLα, which coordinates the binding of other proteins involved with mismatch repair like DNA helicase, single-stranded-DNA binding-protein (RPA), and DNA polymerases.[13][14]
- MSH2 protein dimerizes with MSH6 protein, which identifies mismatches via a sliding clamp model, a protein for scanning for errors.[15][16]
The impairment of either gene for the protein dimer impairs the protein function.[17] These 4 genes are involved in error correction (mismatch repair), so dysfunction of the genes can lead to the inability to fix DNA replication errors and cause HNPCC.[18] HNPCC is known to be associated with other mutations in genes involved in the DNA mismatch repair pathway:
| OMIM name | Genes implicated in HNPCC | Frequency of mutations in HNPCC families | Locus | First publication |
|---|---|---|---|---|
| HNPCC1 ( 120435) | MSH2/EPCAM | approximately 60% | 2p22 | Fishel 1993[16] |
| HNPCC2 ( 609310) | MLH1 | approximately 30% | 3p21 | Papadopoulos 1994[19] |
| HNPCC5 | MSH6 | 7-10% | 2p16 | Miyaki 1997[20] |
| HNPCC4 | PMS2 | relatively infrequent | 7p22[21] | Nicolaides 1994 |
| HNPCC3 | PMS1 | case report[21] | 2q31-q33 | Nicolaides 1994 |
| HNPCC6 | TGFBR2 | case report[22] | 3p22 | |
| HNPCC7 | MLH3 | disputed[23] | 14q24.3 | |
People with MSH6 mutations are more likely to be Amsterdam criteria II-negative.[24] The presentation with MSH6 is slightly different from with MLH1 and MSH2, and the term "MSH6 syndrome" has been used to describe this condition.[25] In one study, the Bethesda guidelines were more sensitive than the Amsterdam Criteria in detecting it.[26]
Up to 39% of families with mutations in an HNPCC gene do not meet the Amsterdam criteria. Therefore, families found to have a deleterious mutation in an HNPCC gene should be considered to have HNPCC regardless of the extent of the family history. This also means that the Amsterdam criteria fail to identify many people who are at risk for Lynch syndrome. Improving the criteria for screening is an active area of research, as detailed in the Screening Strategies section of this article.
Most people with HNPCC inherit the condition from a parent. However, due to incomplete penetrance, variable age of cancer diagnosis, cancer risk reduction, or early death, not all people with an HNPCC gene mutation have a parent who had cancer. Some people develop HNPCC de-novo in a new generation, without inheriting the gene. These people are often only identified after developing an early-life colon cancer. Parents with HNPCC have a 50% chance of passing the genetic mutation on to each child. It is also important to note, that deleterious mutation in one of MMR genes alone is not sufficient to cause cancer, but that rather further mutations in other tumour suppressor genes need to occur.[27]
Diagnosis
A diagnosis of Lynch syndrome is applied to people with a germline DNA mutation in one of the MMR genes (MLH1, MSH2, MSH6, and PMS2) or the EPCAM gene, identified by genetic testing.[28] Candidates for germline genetic testing can be identified by clinical criteria such as the Amsterdam Clinical Criteria and Bethesda Guidelines, or through tumor analysis by immunohistochemistry(IHC), or microsatellite instability (MSI) testing.[28] In the US, professional societies recommend testing every colon cancer for MSI or IHC as screening for Lynch syndrome, but this is not always performed because of cost and resource limitations.[29] Genetic testing is commercially available and consists of a blood test.
Immunohistochemistry
Immunohistochemistry (IHC) is a method that can be used to detect abnormal mismatch repair (MMR) protein expression in tumours that are associated with Lynch syndrome. While it is not diagnostic of a Lynch syndrome, it can play a role in identifying people who should have germline testing.[30] Two methods of implementation of IHC testing includes age-based testing and universal testing for all people.[31] Currently, there is no widespread agreement regarding which screening method should be used.[31] Age-based testing for IHC has been suggested in part due to cost-benefit analyses, whereas universal testing for all people with colorectal cancer ensures people with Lynch Syndrome are not missed.[31] To address the costs, researchers are trying to predict MSI or IHC directly from the way the tumor looks under the microscope, without doing any molecular testing.[29]
Microsatellite Instability
Mutations in DNA mismatch repair systems can lead to difficulty transmitting regions within the DNA which contain repeating patterns of two or three nucleotides (microsatellites), otherwise known as microsatellite instability (MSI).[32] MSI is associated with alternate sized repetitive DNA sequences that are not present in the correlated germ line DNA resulting in 15-20% of colorectal cancers.[33] MSI is identified through DNA extraction from both a tumor tissue sample and a normal tissue sample followed by PCR analysis of microsatellite regions.[32] MSI analysis can be used to identify people who may have Lynch syndrome and direct them for further testing.[32] One study noted that one third of MSI colorectal cancers showed a low immunoscore, suggesting that tumor-infiltrating lymphocytes might be a good option for therapy for these patients. High numbers of tumor-infiltrating lymphocytes were related with better survival rates and treatment responses.[34]
Classification
Three major groups of MSI-H (microsatellite instability – MSI) cancers can be recognized by histopathological criteria:
- right-sided poorly differentiated cancers
- right-sided mucinous cancers
- adenocarcinomas in any location showing any measurable level of intraepithelial lymphocyte (TIL)
The histopathological criteria are not sensitive enough to detect MSI from histology but researchers are trying to use artificial intelligence to predict MSI from histology.[29]
In addition, HNPCC can be divided into Lynch syndrome I (familial colon cancer) and Lynch syndrome II (HNPCC associated with other cancers of the gastrointestinal tract or reproductive system).[35]
Screening
Genetic counseling and genetic testing are recommended for families that meet the Amsterdam criteria, preferably before the onset of colon cancer.
Colon cancer
Colonoscopies are recommended as a preventative method of surveillance for individuals who have Lynch syndrome, or LS-associated genes. Specifically, it is recommended that colonoscopies begin at ages 20–25 for MLH1 and MSH2 mutation carriers and 35 years for MSH6 and PMS2 mutation carriers.[36] Colonoscopic surveillance should then be performed at a 1-2 year interval for Lynch Syndrome patients.[36]
Endometrial/Ovarian cancer
A transvaginal ultrasound with or without endometrial biopsy is recommended annually for ovarian and endometrial cancer screening.[37] For women with Lynch syndrome, a yearly CA-125 blood test can be used to screen for ovarian cancer, however there is limited data on the efficacy of this test in reducing mortality.[38]
Other cancers
There are also strategies for detecting other cancers early or reducing the chances of developing them that people with Lynch syndrome can discuss with their doctor, however their effectiveness is not clear.[39][40] These options include:
- Upper endoscopies to detect stomach and small bowel cancer every 3–5 years, starting at age 30 at the earliest (preferably in a research setting)[39][40]
- Annual urinalysis to detect bladder cancer, starting at age 30 at the earliest (preferably in a research setting)[39][40]
- Annual physical and neurological exams to detect cancer in the central nervous system (brain or spinal cord), starting at age 25 at the earliest[39]
Amsterdam criteria
The following are the Amsterdam criteria in identifying high-risk candidates for molecular genetic testing:[41]
Amsterdam I Criteria (all bullet points must be fulfilled): The Amsterdam I criteria were published in 1990; however, were felt to be insufficiently sensitive.[42]
- Three or more family members with a confirmed diagnosis of colorectal cancer, one of whom is a first degree (parent, child, sibling) relative of the other two
- Two successive affected generations
- One or more colon cancers diagnosed under age 50 years
- Familial adenomatous polyposis (FAP) has been excluded
The Amsterdam II criteria were developed in 1999 and improved the diagnostic sensitivity for Lynch syndrome by including cancers of the endometrium, small bowel, ureter and renal pelvis.[43]
Amsterdam Criteria II (all bullet points must be fulfilled):[43]
- Three or more family members with HNPCC-related cancers, one of whom is a first-degree relative of the other two
- Two successive affected generations
- One or more of the HNPCC-related cancers diagnosed under age 50 years
- Familial adenomatous polyposis (FAP) has been excluded
The Bethesda criteria were developed in 1997 and later updated in 2004 by the National Cancer Institute to identify persons requiring further testing for Lynch syndrome through MSI. In contrast to the Amsterdam Criteria, the Revised Bethesda Guidelines use pathological data in addition to clinical information to help health care providers identify persons at high risk.[42][43]
Revised Bethesda Guidelines
If a person meets any 1 of 5 criteria the tumour(s) from the person should be tested for MSI:[42]
1. Colorectal cancer diagnosed before age 50
2. Presence of synchronous or metachronous colorectal or other Lynch syndrome associated cancers (e.g. cancers of endometrium, ovary, stomach, small bowel, pancreas, biliary tract, ureter, renal pelvis, brain, sebaceous glands, keratoacanthomas)
3. Colorectal cancer with MSI-high pathology in a person who is younger than 60 years of age
4. Colorectal cancer diagnosed in a person with one or more first-degree relative with colorectal cancer or Lynch syndrome associated tumour diagnosed under age 50
5. Person with colorectal cancer and two or more first- or second-degree relatives with colorectal cancer or Lynch syndrome associated cancer diagnosed at any age.[42][43]
It is important to note that these clinical criteria can be difficult to use in practice and clinical criteria used alone misses between 12 and 68 percent of Lynch syndrome cases.[42]
Surgery
Prophylactic hysterectomy and salpingo-oophorectomy (removal of the uterus, Fallopian tubes, and ovaries to prevent cancer from developing) can be performed before ovarian or endometrial cancer develops.[37]
Treatment
Surgery remains the front-line therapy for HNPCC. Patients with Lynch syndrome who develop colorectal cancer may be treated with either a partial colectomy or total colectomy with ileorectal anastomosis. Due to increased risk of colorectal cancer following partial colectomy and similar quality of life after both surgeries, a total colectomy may be a preferred treatment for HNPCC, especially in younger patients.[44]
There is an ongoing controversy over the benefit of 5-fluorouracil-based adjuvant therapies for HNPCC-related colorectal tumours, particularly those in stages I and II.[45]
- Anti-PD-1 antibody therapy can be effective.[46]
Checkpoint blockade with anti-PD-1 therapy is now preferred first line therapy for advanced Microsatellite-Instability–High colorectal cancer.[47]
Epidemiology
Though the exact prevalence of Lynch syndrome-causing mutations in the general population remain unknown, recent studies estimate the prevalence to be 1 in 279 individuals, or 0.35%.[48][49] Certain populations are known to have a higher prevalence of founder mutations, including, but not limited to, French Canadians, Icelanders, African Americans, and Ashkenazi Jews.[48][49] Lynch syndrome-causing mutations are found in approximately 3% of all diagnosed colorectal cancers, and 1.8% of all diagnosed endometrial cancers.[48][49] The average age of diagnosis of cancer in patients with this syndrome is 44 years old, as compared to 64 years old in people without the syndrome.[50]
Terminology
Henry T. Lynch, Professor of Medicine at Creighton University Medical Center, characterized the syndrome in 1966.[51] In his earlier work, he described the disease entity as "cancer family syndrome." The term "Lynch syndrome" was coined in 1984 by other authors; Lynch named the condition HNPCC in 1985. Since then the two terms have been used interchangeably, until later advances in the understanding of the genetics of the disease led to the term HNPCC falling out of favor.[52]
Other sources reserve the term "Lynch syndrome" when there is a known DNA mismatch repair defect, and use the term "familial colorectal cancer type X" when the Amsterdam criteria are met but there is no known DNA mismatch repair defect.[53] The putative "type X" families appear to have a lower overall incidence of cancer and lower risk for non-colorectal cancers than families with documented DNA mismatch repair deficiency.[54] About 35% of people who meet Amsterdam criteria do not have a DNA-mismatch-repair gene mutation.[55]
Complicating matters is the presence of an alternative set of criteria, known as the "Bethesda Guidelines."[56][57][58]
Society
There are a number of non-profit organisations providing information and support, including Lynch Syndrome International, AliveAndKickn, Lynch Syndrome UK[59] and Bowel Cancer UK.[60] In the US, National Lynch Syndrome Awareness Day is March 22.[61]
References
- "Lynch syndrome | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Archived from the original on 16 July 2019. Retrieved 4 October 2019.
- Kastrinos F, Mukherjee B, Tayob N, Wang F, Sparr J, Raymond VM, et al. (October 2009). "Risk of pancreatic cancer in families with Lynch syndrome". JAMA. 302 (16): 1790–5. doi:10.1001/jama.2009.1529. PMC 4091624. PMID 19861671.
- "Lynch Syndrome". DynaMed. February 22, 2019. Retrieved November 18, 2019.
- Vogel JD, Eskicioglu C, Weiser MR, Feingold DL, Steele SR (October 2017). "The American Society of Colon and Rectal Surgeons Clinical Practice Guidelines for the Treatment of Colon Cancer". Diseases of the Colon and Rectum. 60 (10): 999–1017. doi:10.1097/DCR.0000000000000926. PMID 28891842.
- Hoffman BL (2012). "Chapter 33: Endometrial Cancer". Williams Gynecology (2nd ed.). New York: McGraw-Hill Medical. ISBN 978-0071716727. Archived from the original on 2014-01-04. Retrieved 2014-06-23.
- Braun MM, Overbeek-Wager EA, Grumbo RJ (March 2016). "Diagnosis and Management of Endometrial Cancer". American Family Physician. 93 (6): 468–74. PMID 26977831.
- Sobocińska, Joanna; Kolenda, Tomasz; Teresiak, Anna; Badziąg-Leśniak, Natalia; Kopczyńska, Magda; Guglas, Kacper; Przybyła, Anna; Filas, Violetta; Bogajewska-Ryłko, Elżbieta; Lamperska, Katarzyna; Mackiewicz, Andrzej (2020-10-05). "Diagnostics of Mutations in MMR/EPCAM Genes and Their Role in the Treatment and Care of Patients with Lynch Syndrome". Diagnostics. 10 (10): 786. doi:10.3390/diagnostics10100786. ISSN 2075-4418. PMC 7600989. PMID 33027913.
- Møller, Pål; Seppälä, Toni T.; Bernstein, Inge; Holinski-Feder, Elke; Sala, Paulo; Gareth Evans, D.; Lindblom, Annika; Macrae, Finlay; Blanco, Ignacio; Sijmons, Rolf H.; Jeffries, Jacqueline (2018). "Cancer risk and survival in path_MMR carriers by gene and gender up to 75 years of age: a report from the Prospective Lynch Syndrome Database". Gut. 67 (7): 1306–1316. doi:10.1136/gutjnl-2017-314057. ISSN 1468-3288. PMC 6031262. PMID 28754778.
- Ring, Kari L.; Garcia, Christine; Thomas, Martha H.; Modesitt, Susan C. (2017). "Current and future role of genetic screening in gynecologic malignancies". American Journal of Obstetrics and Gynecology. 217 (5): 512–521. doi:10.1016/j.ajog.2017.04.011. ISSN 1097-6868. PMID 28411145. S2CID 29024566.
- "Lynch Syndrome". Genetics Home Reference.
- Pathology of Hereditary Nonpolyposis Colorectal Cancer - JASS 910 (1): 62 - Annals of the New York Academy of Sciences Archived 2006-06-19 at the Wayback Machine
- Oki E, Oda S, Maehara Y, Sugimachi K (March 1999). "Mutated gene-specific phenotypes of dinucleotide repeat instability in human colorectal carcinoma cell lines deficient in DNA mismatch repair". Oncogene. 18 (12): 2143–7. doi:10.1038/sj.onc.1202583. PMID 10321739.
- Yokoyama T, Takehara K, Sugimoto N, Kaneko K, Fujimoto E, Okazawa-Sakai M, et al. (May 2018). "Lynch syndrome-associated endometrial carcinoma with MLH1 germline mutation and MLH1 promoter hypermethylation: a case report and literature review". BMC Cancer. 18 (1): 576. doi:10.1186/s12885-018-4489-0. PMC 5963021. PMID 29783979.
- Peltomäki P (March 2003). "Role of DNA mismatch repair defects in the pathogenesis of human cancer". Journal of Clinical Oncology. 21 (6): 1174–9. doi:10.1200/JCO.2003.04.060. PMID 12637487.
- Tamura K, Kaneda M, Futagawa M, Takeshita M, Kim S, Nakama M, et al. (September 2019). "Genetic and genomic basis of the mismatch repair system involved in Lynch syndrome". International Journal of Clinical Oncology. 24 (9): 999–1011. doi:10.1007/s10147-019-01494-y. PMID 31273487. S2CID 195795805.
- Fishel R, Lescoe MK, Rao MR, Copeland NG, Jenkins NA, Garber J, et al. (December 1993). "The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer". Cell. 75 (5): 1027–38. doi:10.1016/0092-8674(93)90546-3. PMID 8252616.
- Yurgelun MB, Hampel H (May 2018). "Recent Advances in Lynch Syndrome: Diagnosis, Treatment, and Cancer Prevention". American Society of Clinical Oncology Educational Book. American Society of Clinical Oncology. Annual Meeting. 38 (38): 101–109. doi:10.1200/EDBK_208341. PMID 30231390.
- Le S, Ansari U, Mumtaz A, Malik K, Patel P, Doyle A, Khachemoune A (November 2017). "Lynch Syndrome and Muir-Torre Syndrome: An update and review on the genetics, epidemiology, and management of two related disorders". Dermatology Online Journal. 23 (11). doi:10.5070/D32311037239. PMID 29447627.
- Papadopoulos N, Nicolaides NC, Wei YF, Ruben SM, Carter KC, Rosen CA, et al. (March 1994). "Mutation of a mutL homolog in hereditary colon cancer". Science. 263 (5153): 1625–9. Bibcode:1994Sci...263.1625P. doi:10.1126/science.8128251. PMID 8128251.
- Miyaki M, Konishi M, Tanaka K, Kikuchi-Yanoshita R, Muraoka M, Yasuno M, et al. (November 1997). "Germline mutation of MSH6 as the cause of hereditary nonpolyposis colorectal cancer". Nature Genetics. 17 (3): 271–2. doi:10.1038/ng1197-271. PMID 9354786. S2CID 22473295.
- Nicolaides NC, Papadopoulos N, Liu B, Wei YF, Carter KC, Ruben SM, et al. (September 1994). "Mutations of two PMS homologues in hereditary nonpolyposis colon cancer". Nature. 371 (6492): 75–80. Bibcode:1994Natur.371...75N. doi:10.1038/371075a0. PMID 8072530. S2CID 4244907.
- Lu SL, Kawabata M, Imamura T, Akiyama Y, Nomizu T, Miyazono K, Yuasa Y (May 1998). "HNPCC associated with germline mutation in the TGF-beta type II receptor gene". Nature Genetics. 19 (1): 17–8. doi:10.1038/ng0598-17. PMID 9590282. S2CID 46658147.
- Ou J, Rasmussen M, Westers H, Andersen SD, Jager PO, Kooi KA, et al. (April 2009). "Biochemical characterization of MLH3 missense mutations does not reveal an apparent role of MLH3 in Lynch syndrome" (PDF). Genes, Chromosomes & Cancer. 48 (4): 340–50. doi:10.1002/gcc.20644. hdl:11370/b74f7d2b-12fb-4bfc-a8c8-2d8950e81972. PMID 19156873. S2CID 15526044.
- Ramsoekh D, Wagner A, van Leerdam ME, Dinjens WN, Steyerberg EW, Halley DJ, et al. (November 2008). "A high incidence of MSH6 mutations in Amsterdam criteria II-negative families tested in a diagnostic setting". Gut. 57 (11): 1539–44. doi:10.1136/gut.2008.156695. PMID 18625694. S2CID 10608978.
- Suchy J, Lubinski J (June 2008). "MSH6 syndrome". Hereditary Cancer in Clinical Practice. 6 (2): 103–4. doi:10.1186/1897-4287-6-2-103. PMC 2735474. PMID 19804606.
- Goldberg Y, Porat RM, Kedar I, Shochat C, Galinsky D, Hamburger T, et al. (June 2010). "An Ashkenazi founder mutation in the MSH6 gene leading to HNPCC". Familial Cancer. 9 (2): 141–50. doi:10.1007/s10689-009-9298-9. PMID 19851887. S2CID 25479413.
- "Fact Sheet 33 | BOWEL CANCER AND INHERITED PREDISPOSITION". Archived from the original on 2019-02-28.
- Giardiello FM, Allen JI, Axilbund JE, Boland CR, Burke CA, Burt RW, et al. (August 2014). "Guidelines on genetic evaluation and management of Lynch syndrome: a consensus statement by the US Multi-Society Task Force on colorectal cancer". Gastroenterology. 147 (2): 502–26. doi:10.1053/j.gastro.2014.04.001. PMID 25043945.
- Hildebrand, Lindsey A.; Pierce, Colin J.; Dennis, Michael; Paracha, Munizay; Maoz, Asaf (2021-01-21). "Artificial Intelligence for Histology-Based Detection of Microsatellite Instability and Prediction of Response to Immunotherapy in Colorectal Cancer". Cancers. 13 (3): 391. doi:10.3390/cancers13030391. ISSN 2072-6694. PMC 7864494. PMID 33494280.
- Snowsill T, Huxley N, Hoyle M, Jones-Hughes T, Coelho H, Cooper C, et al. (September 2014). "A systematic review and economic evaluation of diagnostic strategies for Lynch syndrome". Health Technology Assessment. 18 (58): 1–406. doi:10.3310/hta18580. PMC 4781313. PMID 25244061.
- Snowsill T, Coelho H, Huxley N, Jones-Hughes T, Briscoe S, Frayling IM, Hyde C (September 2017). "Molecular testing for Lynch syndrome in people with colorectal cancer: systematic reviews and economic evaluation". Health Technology Assessment. 21 (51): 1–238. doi:10.3310/hta21510. PMC 5611555. PMID 28895526.
- Evrard C, Tachon G, Randrian V, Karayan-Tapon L, Tougeron D (October 2019). "Microsatellite Instability: Diagnosis, Heterogeneity, Discordance, and Clinical Impact in Colorectal Cancer". Cancers. 11 (10): 1567. doi:10.3390/cancers11101567. PMC 6826728. PMID 31618962.
- Nouri Nojadeh, Jafar; Behrouz Sharif, Shahin; Sakhinia, Ebrahim (2018). "Microsatellite instability in colorectal cancer" (PDF). EXCLI Journal. 17: 159–168. doi:10.17179/EXCLI2017-948. PMC 5938532. PMID 29743854.
- Taieb, Julien; Svrcek, Magali; Cohen, Romain; Basile, Debora; Tougeron, David; Phelip, Jean-Marc (November 2022). "Deficient mismatch repair/microsatellite unstable colorectal cancer: Diagnosis, prognosis and treatment". European Journal of Cancer. 175: 136–157. doi:10.1016/j.ejca.2022.07.020. PMID 36115290. S2CID 252289107.
- Hereditary Colorectal Cancer Background. From Medscape. By Juan Carlos Munoz and Louis R Lambiase. Updated: Oct 31, 2011
- Monahan KJ, Bradshaw N, Dolwani S, Desouza B, Dunlop MG, East JE, et al. (March 2020). "Guidelines for the management of hereditary colorectal cancer from the British Society of Gastroenterology (BSG)/Association of Coloproctology of Great Britain and Ireland (ACPGBI)/United Kingdom Cancer Genetics Group (UKCGG)". Gut. 69 (3): 411–444. doi:10.1136/gutjnl-2019-319915. PMC 7034349. PMID 31780574.
- Ring KL, Garcia C, Thomas MH, Modesitt SC (November 2017). "Current and future role of genetic screening in gynecologic malignancies". American Journal of Obstetrics and Gynecology. 217 (5): 512–521. doi:10.1016/j.ajog.2017.04.011. PMID 28411145. S2CID 29024566.
- Sroczynski G, Gogollari A, Conrads-Frank A, Hallsson LR, Pashayan N, Widschwendter M, Siebert U (July 2020). "Cost-Effectiveness of Early Detection and Prevention Strategies for Endometrial Cancer-A Systematic Review". Cancers. 12 (7): 1874. doi:10.3390/cancers12071874. PMC 7408795. PMID 32664613.
- "Medical Options | CDC". www.cdc.gov. 2020-04-01. Retrieved 2020-12-07.
- Vasen, Hans F A; Blanco, Ignacio; Aktan-Collan, Katja; Gopie, Jessica P; Alonso, Angel; Aretz, Stefan; Bernstein, Inge; Bertario, Lucio; Burn, John; Capella, Gabriel; Colas, Chrystelle (June 2013). "Revised guidelines for the clinical management of Lynch syndrome (HNPCC): recommendations by a group of European experts". Gut. 62 (6): 812–823. doi:10.1136/gutjnl-2012-304356. ISSN 0017-5749. PMC 3647358. PMID 23408351.
- Vasen HF, Watson P, Mecklin JP, Lynch HT (June 1999). "New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC". Gastroenterology. 116 (6): 1453–6. doi:10.1016/S0016-5085(99)70510-X. PMID 10348829.
- Vindigni SM, Kaz AM (April 2016). "Universal Screening of Colorectal Cancers for Lynch Syndrome: Challenges and Opportunities". Digestive Diseases and Sciences. 61 (4): 969–76. doi:10.1007/s10620-015-3964-6. PMID 26602911. S2CID 6014333.
- Bui QM, Lin D, Ho W (February 2017). "Approach to Lynch Syndrome for the Gastroenterologist". Digestive Diseases and Sciences. 62 (2): 299–304. doi:10.1007/s10620-016-4346-4. PMID 27990589. S2CID 32833106.
- Stjepanovic N, Moreira L, Carneiro F, Balaguer F, Cervantes A, Balmaña J, Martinelli E; ESMO Guidelines Committee. Hereditary gastrointestinal cancers: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow- up†. Ann Oncol. 2019 Oct 1;30(10):1558-1571. doi: 10.1093/annonc/mdz233. PMID 31378807.
- Boland CR, Koi M, Chang DK, Carethers JM (2007). "The biochemical basis of microsatellite instability and abnormal immunohistochemistry and clinical behavior in Lynch syndrome: from bench to bedside". Familial Cancer. 7 (1): 41–52. doi:10.1007/s10689-007-9145-9. PMC 2847875. PMID 17636426.
- Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. (June 2015). "PD-1 Blockade in Tumors with Mismatch-Repair Deficiency". The New England Journal of Medicine. 372 (26): 2509–20. doi:10.1056/NEJMoa1500596. PMC 4481136. PMID 26028255.
- André, Thierry; Shiu, Kai-Keen; Kim, Tae Won; Jensen, Benny Vittrup; Jensen, Lars Henrik; Punt, Cornelis; Smith, Denis; Garcia-Carbonero, Rocio; Benavides, Manuel; Gibbs, Peter; de la Fouchardiere, Christelle; Rivera, Fernando; Elez, Elena; Bendell, Johanna; Le, Dung T.; Yoshino, Takayuki; Van Cutsem, Eric; Yang, Ping; Farooqui, Mohammed Z.H.; Marinello, Patricia; Diaz, Luis A. (3 December 2020). "Pembrolizumab in Microsatellite-Instability–High Advanced Colorectal Cancer". New England Journal of Medicine. 383 (23): 2207–2218. doi:10.1056/NEJMoa2017699. PMID 33264544. S2CID 227259533.
- Boland PM, Yurgelun MB, Boland CR (May 2018). "Recent progress in Lynch syndrome and other familial colorectal cancer syndromes". CA: A Cancer Journal for Clinicians. 68 (3): 217–231. doi:10.3322/caac.21448. PMC 5980692. PMID 29485237.
- Biller LH, Syngal S, Yurgelun MB (April 2019). "Recent advances in Lynch syndrome". Familial Cancer. 18 (2): 211–219. doi:10.1007/s10689-018-00117-1. PMC 6450737. PMID 30627969.
- "Oncolink". www.oncolink.org. Archived from the original on July 22, 2011.
- Lynch HT, Shaw MW, Magnuson CW, Larsen AL, Krush AJ (February 1966). "Hereditary factors in cancer. Study of two large midwestern kindreds". Archives of Internal Medicine. 117 (2): 206–12. doi:10.1001/archinte.117.2.206. PMID 5901552.
- Bellizzi AM, Frankel WL (November 2009). "Colorectal cancer due to deficiency in DNA mismatch repair function: a review". Advances in Anatomic Pathology. 16 (6): 405–17. doi:10.1097/PAP.0b013e3181bb6bdc. PMID 19851131. S2CID 25600795.
- Lindor NM (October 2009). "Familial colorectal cancer type X: the other half of hereditary nonpolyposis colon cancer syndrome". Surgical Oncology Clinics of North America. 18 (4): 637–45. doi:10.1016/j.soc.2009.07.003. PMC 3454516. PMID 19793571.
- Lindor NM, Rabe K, Petersen GM, Haile R, Casey G, Baron J, et al. (April 2005). "Lower cancer incidence in Amsterdam-I criteria families without mismatch repair deficiency: familial colorectal cancer type X". JAMA. 293 (16): 1979–85. doi:10.1001/jama.293.16.1979. PMC 2933042. PMID 15855431.
- Scott RJ, McPhillips M, Meldrum CJ, Fitzgerald PE, Adams K, Spigelman AD, et al. (January 2001). "Hereditary nonpolyposis colorectal cancer in 95 families: differences and similarities between mutation-positive and mutation-negative kindreds". American Journal of Human Genetics. 68 (1): 118–127. doi:10.1086/316942. PMC 1234904. PMID 11112663.
- Gologan A, Krasinskas A, Hunt J, Thull DL, Farkas L, Sepulveda AR (November 2005). "Performance of the revised Bethesda guidelines for identification of colorectal carcinomas with a high level of microsatellite instability". Archives of Pathology & Laboratory Medicine. 129 (11): 1390–7. doi:10.5858/2005-129-1390-POTRBG. PMID 16253017.
- Umar A, Boland CR, Terdiman JP, Syngal S, de la Chapelle A, Rüschoff J, et al. (February 2004). "Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability". Journal of the National Cancer Institute. 96 (4): 261–8. doi:10.1093/jnci/djh034. PMC 2933058. PMID 14970275.
- Lipton LR, Johnson V, Cummings C, Fisher S, Risby P, Eftekhar Sadat AT, et al. (December 2004). "Refining the Amsterdam Criteria and Bethesda Guidelines: testing algorithms for the prediction of mismatch repair mutation status in the familial cancer clinic". Journal of Clinical Oncology. 22 (24): 4934–43. doi:10.1200/JCO.2004.11.084. PMID 15611508. Archived from the original on 2013-04-15.
- "Lynch Syndrome UK". Retrieved 31 March 2018.
- "Bowel Cancer UK: Lynch Syndrome". Retrieved 31 March 2018.
- "CDC: March 22nd is National Lynch Syndrome Awareness Day!". 2018-03-20. Retrieved 31 March 2018.
Further reading
Books
- McKay, Ami (2019). Daughter of Family G. : a memoir of cancer genes, love and fate. Toronto: Alfred A. Knopf Canada. ISBN 978-0-345-80946-9. OCLC 1089450897. Paperback version retitled Before My Time.