Hereditary spherocytosis
Hereditary spherocytosis (HS) is a congenital hemolytic disorder, wherein a genetic mutation coding for a structural membrane protein phenotype leads to a spherical shaping of erythrocytic cellular morphology. As erythrocytes are sphere-shaped (spherocytosis), rather than the normal biconcave disk-shaped, their morphology interferes with these cells' abilities to be flexible during circulation throughout the entirety of the body - arteries, arterioles, capillaries, venules, veins, and organs. This difference in shape also makes the red blood cells more prone to rupture[1] under osmotic and/or mechanical stress. Cells with these dysfunctional proteins are degraded in the spleen, which leads to a shortage of erythrocytes resulting in hemolytic anemia.
| Hereditary spherocytosis | |
|---|---|
| Other names | Minkowski–Chauffard syndrome |
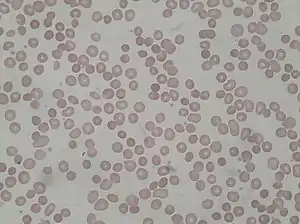 | |
| Peripheral blood smear from patient with hereditary spherocytosis | |
| Specialty | Hematology |
HS was first described in 1871, and is the most common cause of inherited hemolysis in populations of northern European descent, with an incidence of 1 in 5000 births. The clinical severity of HS varies from mild (symptom-free carrier), to moderate (anemic, jaundiced, and with splenomegaly), to severe (hemolytic crisis, in-utero hydrops fetalis), because HS is caused by genetic mutations in a multitude of structural membrane proteins and exhibits incomplete penetrance in its expression.
Early symptoms include anemia, jaundice, splenomegaly, and fatigue.[2] Acute cases can threaten to cause hypoxia secondary to anemia and acute kernicterus through high blood levels of bilirubin, particularly in newborns. Most cases can be detected soon after birth. Testing for HS is available for the children of affected adults. Occasionally, the disease will go unnoticed until the child is about 4 or 5 years of age. A person may also be a carrier of the disease and show no signs or symptoms of the disease. Late complications may result in the development of pigmented gallstones, which is secondary to the detritus of the broken-down blood cells (unconjugated or indirect bilirubin) accumulating within the gallbladder. Also, patients who are heterozygous for a hemochromatosis gene may exhibit iron overload, despite the hemochromatosis genes being recessive.[3][4] In chronic patients, an infection or other illness can cause an increase in the destruction of red blood cells, resulting in the appearance of acute symptoms, a hemolytic crisis. On a blood smear, Howell-Jolly bodies may be seen within red blood cells. Primary treatment for patients with symptomatic HS has been total splenectomy, which eliminates the hemolytic process, allowing normal hemoglobin, reticulocyte and bilirubin levels. The resultant asplenic patient is susceptible to encapsulated bacterial infection, and prevented with vaccination. If other symptoms, such as abdominal pain persist, the removal of the gallbladder may be warranted for symptomatic cholelithiasis.
Epidemiology
Hereditary spherocytosis is the heritable hemolytic disorder, affecting 1 in 2,000 people of Northern European ancestry.[5] According to Harrison's Principles of Internal Medicine, the frequency is at least 1 in 5,000 within the United States of America.[6] While HS is most commonly (though not exclusively) found in Northern European and Japanese families, an estimated 25% of cases are due to spontaneous mutations.
Etiology
Hereditary spherocytosis is an erythrocytic disorder of that affects the following red cell membrane proteins in a congenital fashion:
- Spectrin (alpha and beta)[7]
- Ankyrin[7]
- Band-3 Protein[8]
- Protein-4.2[8]
- Lesser proteins of significance[6]
Hereditary spherocytosis can be an autosomal recessive or autosomal dominant trait.[9] The autosomal recessive inheritance pattern accounts for close to 25% of the clinical cases. The autosomal dominant inheritance pattern accounts for over 75% of the clinical cases. Many positive individuals will not present clinically, thus the etiologic data may be artificially skewed towards the more prominent dominant forms. These dominant forms tend to leave a family history that yields generational splenectomies and black gallstones cholelithiasis. Lastly, an estimated 25% of cases are due to spontaneous mutations.
Pathophysiology
Causative Genetic Mutations and Phenotypic Expressions
Hereditary spherocytosis is caused by a variety of molecular defects in the genes that code for the red blood cell proteins spectrin (alpha and beta), ankyrin,[7] band 3 protein, protein 4.2,[8] and other red blood cell membrane proteins:[6]
| Hereditary Spherocytosis Type | Genotypic Etiology | Phenotypic Expression | ||
|---|---|---|---|---|
| OMIM* | Gene | Locus | Erythrocyte Membrane Protein Affected | |
| HS-1 | 182900 | ANK1 | 8p11.2 | Ankyrin |
| HS-2 | 182870 | SPTB | 14q22-q23 | Spectrin (Beta)* |
| HS-3 | 270970 | SPTA | 1q21 | Spectrin (Alpha-1)* |
| HS-4 | 612653 | SLC4A1 | 17q21-q22 | Band-3 Protein |
| HS-5 | 612690 | EPB42 | 15q15 | Protein-4.2 |
*Online Mendelian Inheritance in Man (OMIM). The Alpha-1 refers the Alpha-1 Subunit of the Spectrin protein. The Beta refers the Beta Subunit of the Spectrin protein.
These genetic mutations are acted upon and executed by erythrocyte progenitor cells within the bone marrow, where red blood cells are normally produced in the non-pathological state (see Extramedullary Hematopoiesis for pathological production outside of the bone marrow).
Pathophysiology of Mutated Erythrocytic Membrane Proteins
These proteins are necessary to maintain the normal shape of a red blood cell, which is a biconcave disk. The integrating protein that is most commonly defective is spectrin which is responsible for incorporation and binding of spectrin to the greater actin cytoskeleton. This dysfunction of cytoskeletal instabilities ensue, and leave the plasma membrane of the cell less supported and/or weakened.
| Erythrocyte Membrane Protein | Protein Function Compromised | Process Effect | Pathogenesis of HS | Likelihood | Mechanical Effect
(In Order) |
|---|---|---|---|---|---|
| Spectrin (Alpha-1) | Alpha-1 subunit Actin Association | Failure in plasma membrane tethering to actin cytoskeleton. | 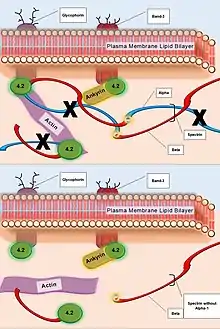 Caption: The deficiency of Alpha-1 subunits in Spectrin protein network (top). The resulting failed or weakened tethering of Spectrin interior to the plasma membrane to the greater actin cytoskeleton (bottom). |
Most Common |
 Micrograph of a spherocyte (center). |
| Spectrin (Beta) | Hydrophobic interactions and electrostatic attraction to ankyrin and actin | Failure in plasma membrane tethering to actin cytoskeleton. | 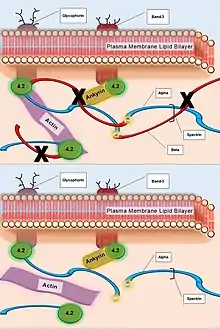 Caption: The deficiency of Beta subunits in Spectrin protein network (top). The resulting failed or weakened tethering of Spectrin interior to the plasma membrane to the greater actin cytoskeleton (bottom). | ||
| Ankyrin | Hydrophobic interactions and electrostatic attraction to Beta subunit | Failure to mediate anchorage of integral plasma membrane proteins to spectrin. | 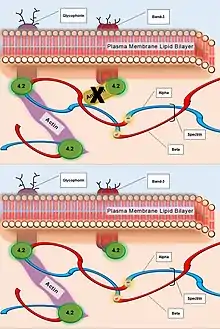 Caption: The deficiency of Ankyrin that normally associates with plasma membrane proteins (top). The resulting failed or weakened anchorage of Spectin to interior plasma membrane, and subsequently weakened association between the greater actin cytoskeleton and the plasma membrane (bottom). |
Common | |
| Band-3 | Failure to mediate the exchange of chloride (Cl−) with bicarbonate (HCO3−) across plasma membranes. | Deficiency causes reduced lipid-stabilization at the plasma membrane. | 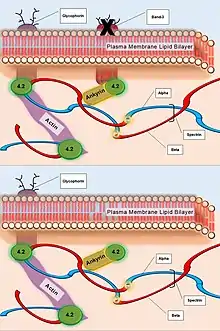 Caption: The deficiency of Band-3 Protein (top). The resulting destabilized plasma membrane without integral Band-3 Protein for association with Protein-4.2 and subsequently Ankyrin (bottom). |
Less Common | |
| Protein-4.2 | Failure of ATP binding that regulates the association of Band-3 with Ankyrin. | Reduced density of plasma cell membrane skeleton, destabilizing the plasma lipid bilayer, thus releasing Band-3 protein-containing microvesicles. | 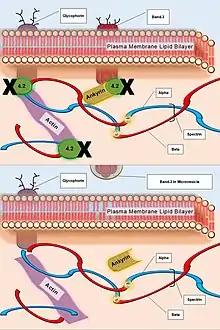 Caption: The deficiency of Protein-4.2 (top). The resulting inability of the greater actin cytoskeleton to associate with the plasma membrane via Spectrin and Ankyrin, and the subsequent loss of Band-3 Protein within microvesicles (bottom). |
Least Common |
Cellular Implications
A secondary defect in hereditary spherocytosis is a deficiency of membrane surface area. The decrease in surface area leads to less efficient gas exchange of the erythrocyte at the alveoli of the lungs and throughout circulation. Decreased surface area may be produced by two different mechanisms:
- Defects of Spectrin, Ankyrin (most commonly), or Protein 4.2 lead to reduced structural integrity of the plasma membrane, destabilizing the overlying lipid bilayer, and releasing band 3-containing microvesicles. Band-3 is important for gas exchange (as seen above).
- Defects of band 3 lead to band 3 deficiency and loss of its lipid-stabilizing effect within the plasma membrane lipid bilayer. This results in the release of band 3-free microvesicles.
Both pathways result in compromised plasma membrane integrity, decreased surface area, and formation of spherocytes with decreased mechanical compliance during circulation.
Cardiovascular and Organ Sequelae
Spherocytes have less plasma membrane compliance and fluidity, and this has implications throughout the entirety of circulation within the body, i.e. arteries, arterioles, capillaries, venules, veins, and organs. The lack of compliance and fluidity lead to a higher viscosity value for blood (thickened blood), which can have implications for both larger and smaller diameter vasculature. However, the most pronounced issues with the lack of compliance and fluidity declare themselves in the failure of the erythrocyte to deform itself when transiting arterioles, capillary beds, and venules. These vessels are smaller, and can become congested or blocked altogether. Studies have demonstrated that HS is related to deep vein thrombosis (DVT) and arterial cardiovascular disease for this reason.
The spleen typically acts as a filter for blood, and targets pathogens and other damaged cells within circulation. Removing blood contaminants promotes entire-body homeostasis. The following facts about the spleen's role in normal body functioning are crucial to understanding the implications of HS on the overall health of the individual:
- Removal of pathogens includes encapsulated bacteria, which reduces sepsis risk (see Complications below).
- Removal of damaged erythrocytes reduces iron burden from heme via hepatosplenic recycling, which reduces sepsis risk (see Complications below).
- Removal of damaged erythrocytes continues healthy signaling to bone marrow, where erythropoiesis replaces red blood cells lost to wear and tear.
In relation to the three facts above, the spleen misguidedly filters spherocytes - regardless of the age or functional status of the cell. The spleen is not dysfunctional itself when selecting spherocytes for destruction, as this is the regular function of the spleen both at an anatomic structure level, and at the molecular cellular level. At the anatomic level, the passage from the Cords of Billroth into the Sinusoids may be seen as a "bottleneck", where red blood cells need to be flexible in order to pass through. In HS, the erythrocytes fail to pass through fenestrations, and this is where resident splenic macrophages sample, or "bite," part of the stuck spherocytes' plasma membranes. The macrophages recognize that the spherocyte is not normal, and the macrophage is "programmed" to destroy irregularities filtered at the spleen from the blood. The resident splenic macrophages therefore phagocytose the spherocytes, causing extravascular hemolysis.[10] This leads to both splenomegaly and anemia. Should this process continue unchecked chronically, inappropriate regulation of erythropoeisis leads to extramedullary hematopoiesis.
Clinical Presentation
HS patients present in a vast array of presentations from being asymptomatic, to the extreme situations of splenic rupture and/or hemolytic crisis, or in-utero demise.
- Asymptomatic HS (mild): 20-30% of patients.
- Infantile-Onset HS (moderate): 60-75% of patients.
- Neonatal or In-Utero Onset HS (severe): <5% of patients.
The most common presentation will demonstrate jaundice (due to increased unconjugated bilirubin), anemia (with secondary pallor), and a palpable spleen sometimes with concomitant tenderness (due to splenic congestion and splenomegaly). Its worth noting that a subsection of HS patients will also have incidental black pigmented gallstones (made of calcium bilirubinate consequence of the extravascular hemolysis), and some of these patients will develop cholelithiasis and/or the potential complex sequelae of this condition, i.e. cholecystisis, choledocholithiasis, etc.
Diagnostics
Laboratory Testing
Available lab testing that may aid in the diagnosis of HS is as follows:
- Peripheral Blood Smear
- Supportive blood work: mean cell volume (MCV), mean corpuscular hemoglobin concentration (MCHC), red blood cell distribution width (RDW), red blood cell count (RBC), reticulocytes, unconjugated bilirubin, haptoglobin, lactate dehydrogenase (LDH).
- Eosin-5-maleimide Binding Test
- Osmotic Fragility Test[11]
- Acidified Glycerol Lysis Test
- A negative direct antiglobin test (Coombs test)
Common Laboratory Findings
The common findings of lab testing in setting of a patient with hereditary spherocytosis:
- Coombs Test: Negative (rules-out autoimmune hemolytic anemia)
- Osmotic Fragility Test: Positive (Spherocytes will rupture in liquid solutions less concentrated than the inside of the red blood cell. This is due to increased permeability of the spherocyte membrane to salt and water, which enters the concentrated inner environment of the RBC and leads to its rupture.[12] Although the osmotic fragility test is no longer considered the gold standard for diagnosing hereditary spherocytosis, as it misses ~25% of cases).
- Acidified Glycerol Lysis Test: Positive (A newer version of the osmotic fragility test that adds glycerol to a hypotonic solution. This produces lysis, a positive test, quicker that the traditional saline version).
- Supportive blood work:
- Mean Cell Volume (MCV): Normocytic (normal range: 80-100fL), or slightly lower. Spherocytes are slightly smaller than normal biconcave red blood cells.
- Mean Corpuscular Hemoglobin Concentration (MCHC): Increased (normal range: 31-36% Hb/cell). This is secondary to less water being in the cell.
- Red Blood Cell Distribution Width (RDW): Increased (normal range: 11-15%). The spherocytes create variation in the size of the red blood cells on average, thus expanding the distribution.
- Red Blood Cell Count (RBC): Sometimes Increased Early (normal range males: 4.3-5.9 million/mm3; normal range females: 3.5-5.5million/mm3). The loss of surface area per cells cause the body to mass produce red blood cells. The spleen will filter spherocytes out and change this value.
- Reticulocytes: Increased (normal range: 0.5%-1.5% of the RBC listed above). The body to mass produces red blood cells (reticulocytes being young erythrocytes) even as the spleen filters spherocytes out. This is known as reticulocytosis.
- Unconjugated Bilirubin: Increased (normal range: 0.2-1.2 mg/dL). This is caused by heme released into the hepatosplenic circulation by macrophages that have phagocytosed erythrocytes. The unconjugated bilirubin is not soluble in water (blood), so it binds to albumin, and is processed in the liver.
- Haptoglobin (Free): Decreased (normal range: 41–165 mg/dL). This is caused by hemoglobin binding to haptoglobin, thus making it no longer "free."
- lactate dehydrogenase (LDH): Increased (Normal range: 110-295U/L in children). This is due to extravascular hemolysis.
- Peripheral Blood Smear: Directly shows spherocytes on microscope.
- Eosin-5-maleimide Binding Test: Positive (reduced mean fluorescence), as the test will demonstrate a reduced ability of the Eosin-5-maleimide dye to bind to erythrocyte plasma membrane proteins. The process relies upon flow cytometry. Gold standard test that produces results at low cost within ~2 hours.
In chronic cases, patients who have taken iron supplementation, have heterozygous hemochromatosis, or received numerous blood transfusions, iron overload may cause additional health issues. Measuring iron stores is sometimes considered part of the diagnostic approach to hereditary spherocytosis in older patients presenting with heart muscle damage of unknown etiology or liver disease without apparent cause.
Imaging
Ultrasound is often used to evaluate the dimensions of the spleen, and also the gallbladder in preparation for functionally curative splenectomy with or without cholecystectomy.
Treatment
Although research is ongoing, currently there is no genetic-level cure for the myriad of mutations that cause the various presentations of hereditary spherocytosis.[6] Common current management focuses on interventions prevent the body from inappropriately destroying the functional spherocytes produced by erythrocyte progenitor cells within the bone marrow.
Typical treatment options include:
- Splenectomy (operative): This is the operative removal of the entire spleen by a surgeon, thus stopping the extravascular hemolysis, but also removing the immune functions the spleen naturally provide to the body.
- Partial splenectomy (operative): This is when only a section of the spleen is removed, instead of the entire organ. The goal is to lower the extravascular hemolysis to a level compatible with homeostasis of the patient, while preserving the immune function of the spleen's presence.[2] Research on outcomes is currently ongoing,[2] and is routine at children's hospitals within the United States of America.[13]
- Splenic ablation (interventional radiology): This option is non-operative, and is when an interventional radiologist (instead of a surgeon) uses radiologic techniques to coil or cauterize vasculature within the splenic circulation. There are various points in the circulatory pathway where the treatment can be applied to produces varying amounts of retained viability of the spleen, thus giving the option to attempt to preserve splenic immune function.
All operative and interventional treaments require the immunization of HS patients against the influenza virus, SARS-CoV-2, and encapsulated bacteria such as Streptococcus pneumoniae and meningococcus. Antibiotics are no longer recommended for maintenance use, even in post-splenectomy HS patients. Since the spleen is important for protecting against encapsulated organisms, sepsis caused by encapsulated organisms is a possible complication of splenectomy.[2]
Additional elective treatments offered:
Experimental treatment:
- Bone marrow transplant (heme-oncology intervention): The replacement of the bone marrow with bone marrow from an individual without spherocytosis. The transplanted progenitor cells do not have the genetic mutations found in individuals with hereditary spherocytosis, and therefore do not produce spherocytes. This results in an individual with biconcave-disc-shaped erythrocytes. This treatment is not standard of care, and is not offered as it has only been documented incidentally during treatment for other diseases, such as myelodysplastic sydnrome.
Symptomatic treatments:
- Folic acid supplementation.
Complications
Common Complications
- Hemolytic crisis, with more pronounced jaundice due to accelerated hemolysis (may be precipitated by infection).
- Aplastic crisis with dramatic fall in hemoglobin level and (reticulocyte count)-decompensation, usually due to maturation arrest and often associated with megaloblastic changes; may be precipitated by infection, such as influenza, notably with parvovirus B19.
- Folate deficiency caused by increased bone marrow requirement.
- Pigmented gallstones occur in approximately half of untreated patients. Increased hemolysis of red blood cells leads to increased bilirubin levels, because bilirubin is a breakdown product of heme. The high levels of bilirubin must be excreted into the bile by the liver, which may cause the formation of a pigmented gallstone, which is composed of calcium bilirubinate. Since these stones contain high levels of calcium carbonates and phosphate, they are radiopaque and are visible on x-ray.
- Hyperglycemia with concomitant low hemoglobin A1C levels.
- Hemoglobin A1C (glycated hemoglobin) is a test for determining the average blood glucose levels over an extended period of time, and is often used to evaluate glucose control in diabetics. The hemoglobin A1C levels are abnormally low because the life span of the red blood cells is decreased - providing less time for the non-enzymatic glycosylation of hemoglobin. Thus, even with high overall blood sugar, the A1C will be lower than expected.
- Iron overload.
- Leg ulcer.
- Deep vein thrombosis (DVT).
- Cardiovascular disease.
Research
Modern ongoing research interests:
- Experimental gene therapy exists to treat hereditary spherocytosis in lab mice; however, this treatment has not yet been tried on humans due to all of the risks involved in human gene therapy.
- Bone marrow transplant.
- Paradoxical endurance-based athleticism associated with hereditary spherocytosis.
- Evolution of spherocytic erythrocyte adaptation in those of Northern European and Japanese descent.
- Increased ability to defend against viral infections in hereditary spherocytosis patients.
See also
References
- Cotran, Ramzi S.; Kumar, Vinay; Fausto, Nelson; Nelso Fausto; Robbins, Stanley L.; Abbas, Abul K. (2005). Robbins and Cotran pathologic basis of disease. St. Louis, Mo: Elsevier Saunders. p. 625. ISBN 0-7216-0187-1.
- Bolton-Maggs, P. H. B.; Stevens, R. F.; Dodd, N. J.; Lamont, G.; Tittensor, P.; King, M. -J.; General Haematology Task Force of the British Committee for Standards in Haematology (2004). "Guidelines for the diagnosis and management of hereditary spherocytosis". British Journal of Haematology. 126 (4): 455–474. doi:10.1111/j.1365-2141.2004.05052.x. PMID 15287938.
- J L Rasmussen; D A Odelson; F L Macrina (1987-08-01). "Complete nucleotide sequence of insertion element IS4351 from Bacteroides fragilis. - UKPMC Article - UK PubMed Central". UKPMC Article. Archived from the original on 2012-12-23. Retrieved 2012-07-03.
- Paula Bolton-Maggs (September 2011). "Guidelines for the Diagnosis and Management of Hereditary Spherocytosis" (PDF). The British Committee for Standards in Haematology. Archived from the original (PDF) on 12 May 2012. Retrieved 2 July 2012.
- "Hereditary spherocytosis: MedlinePlus Genetics".
- Anthony S. Fauci; Eugene Braunwald; Dennis L. Kasper; Stephen L. Hauser; Dan L. Longo; J. Larry Jameson; Joseph Loscalzo (2008). Harrison's principles of internal medicine (17th ed.). New York: McGraw-Hill Medical. pp. Chapter 106. ISBN 978-0071466332.
- Gallagher PG, Forget BG (December 1998). "Hematologically important mutations: spectrin and ankyrin variants in hereditary spherocytosis". Blood Cells Mol. Dis. 24 (4): 539–43. doi:10.1006/bcmd.1998.0217. PMID 9887280.
- Perrotta S, Gallagher PG, Mohandas N (October 2008). "Hereditary spherocytosis". Lancet. 372 (9647): 1411–26. doi:10.1016/S0140-6736(08)61588-3. PMID 18940465. S2CID 10926437.
- Eber S, Lux SE (April 2004). "Hereditary spherocytosis--defects in proteins that connect the membrane skeleton to the lipid bilayer". Semin. Hematol. 41 (2): 118–41. doi:10.1053/j.seminhematol.2004.01.002. PMID 15071790.
- Mitchell, Richard Sheppard; Kumar, Vinay; Abbas, Abul K.; Fausto, Nelson (2007). "12". Robbins Basic Pathology. Philadelphia: Saunders. p. 425. ISBN 978-1-4160-2973-1. 8th edition.
- Won DI, Suh JS (March 2009). "Flow cytometric detection of erythrocyte osmotic fragility". Cytometry Part B. 76 (2): 135–41. doi:10.1002/cyto.b.20448. PMID 18727072. S2CID 7339411.
- Goljan. Rapid Review Pathology. 2010. Page 213.
- Buesing, K. L.; Tracy, E. T.; Kiernan, C.; Pastor, A. C.; Cassidy, L. D.; Scott, J. P.; Ware, R. E.; Davidoff, A. M.; Rescorla, F. J.; Langer, J. C.; Rice, H. E.; Oldham, K. T. (2011). "Partial splenectomy for hereditary spherocytosis: A multi-institutional review". Journal of Pediatric Surgery. 46 (1): 178–183. doi:10.1016/j.jpedsurg.2010.09.090. PMID 21238662.