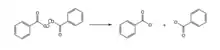
Homolysis (chemistry)
In chemistry, homolysis (from Greek ὅμοιος (homoios) 'equal', and λύσις (lusis) 'loosening') or homolytic fission is the dissociation of a molecular bond by a process where each of the fragments (an atom or molecule) retains one of the originally bonded electrons. During homolytic fission of a neutral molecule with an even number of electrons, two free radicals will be generated.[1] That is, the two electrons involved in the original bond are distributed between the two fragment species. Bond cleavage is also possible by a process called heterolysis.
![]()
The energy involved in this process is called bond dissociation energy (BDE).[2] BDE is defined as the "enthalpy (per mole) required to break a given bond of some specific molecular entity by homolysis," symbolized as D.[3] BDE is dependent on the strength of the bond, which is determined by factors relating to the stability of the resulting radical species.
Because of the relatively high energy required to break bonds in this manner, homolysis occurs primarily under certain circumstances:
- Light (i.e. ultraviolet radiation)
- Heat
- Certain intramolecular bonds, such as the O–O bond of a peroxide, are weak enough to spontaneously homolytically dissociate with a small amount of heat.
- High temperatures in the absence of oxygen (pyrolysis) can induce homolytic elimination of carbon compounds.[4]
- Most bonds homolyse at temperatures above 200°C. [5]
Additionally, in some cases pressure can induce the formation of radicals.[6] These conditions excite electrons to the next highest molecular orbital, thus creating a singly occupied molecular orbital (SOMO).
Adenosylcobalamin is the cofactor which creates the deoxyadenosyl radical by homolytic cleavage of a cobalt-carbon bond in reactions catalysed by methylmalonyl-CoA mutase, isobutyryl-CoA mutase and related enzymes. This triggers rearrangement reactions in the carbon framework of the substrates on which the enzymes act.[7]
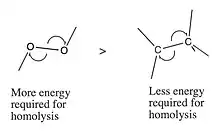
Factors that drive homolysis
Homolytic cleavage is driven by the ability of a molecule to absorb energy from light or heat, and the bond dissociation energy (enthalpy). If the radical species is better able to stabilize the free radical, the energy of the SOMO will be lowered, as will the bond dissociation energy. Bond dissociation energy is determined by multiple factors:[5]
- Electronegativity
- Less electronegative atoms are better stabilizers of radicals, meaning that a bond between two electronegative atoms will have a higher BDE than a similar molecule with two less electronegative atoms. [5]
- Polarizability
- The larger the electron cloud, the better an atom can stabilize the radical (i.e. Iodine is very polarizable and a radical stabilizer). [5]

- Orbital hybridization
- The s-character of an orbital relates to how close electrons are to the nucleus. In the case of a radical, s-character more specifically relates to how close the single electron is to the nucleus. Radicals decrease in stability as they are closer to the nucleus, because the electron affinity of the orbital increases. As a general rule, hybridizations minimizing s-character increase the stability of radicals, and decreases the bond dissociation energy (i.e. sp3 hybridization is most stabilizing).[8]
- Resonance
- Radicals can be stabilized by the donation of negative charge from resonance, or in other words, electron delocalization.
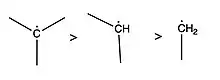
- Hyperconjugation
- Carbon radicals are stabilized by hyperconjugation, meaning that more substituted carbons are more stable, and hence have lower BDEs.
- In 2005, Gronert proposed an alternative hypothesis involving the relief of substituent group steric strain (as opposed to the before accepted paradigm, which suggests that carbon radicals are stabilized via alkyl groups). [10]
- The captodative effect
- Radicals can be stabilized by a synergistic effect of both electron-withdrawing group and electron-donating group substituents.
- Electron-withdrawing groups often contain empty π* orbitals that are low in energy and overlap with the SOMO, creating two new orbitals: one that is lower in energy and stabilizing to the radical, and an empty higher energy orbital. Similarly, electron-donating orbitals combine with the radical SOMO, allowing a lone pair to lower in energy and the radical to enter the new higher energy orbital. This interaction is net stabilizing. [5]
See also
References
- IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "homolysis (homolytic)". doi:10.1351/goldbook.H02851
- St. John, P.C., Guan, Y., Kim, Y. et al. Prediction of organic homolytic bond dissociation enthalpies at near chemical accuracy with sub-second computational cost. Nat Commun 11, 2328 (2020). https://doi.org/10.1038/s41467-020-16201-z
- IUPAC. Compendium of Chemical Terminology, 2nd ed. (the "Gold Book"). Compiled by A. D. McNaught and A. Wilkinson. Blackwell Scientific Publications, Oxford (1997). Online version (2019-) created by S. J. Chalk. ISBN 0-9678550-9-8. https://doi.org/10.1351/goldbook.
- I. Pastorova, "Cellulose Char Structure: a Combined Analytical Py-GC-MS, FTIR, and NMR Study", Carbohydrate Research, 262 (1994) 27-47.
- Clayden, Jonathan, Greeves, Nick, Warren, Stuart. (2012). Organic Chemistry (Second ed.). Oxford: OUP. ISBN 978-0-19-927029-3
- Kristina Lekin, Hoa Phan, Stephen M. Winter, Joanne W. L. Wong, Alicea A. Leitch, Dominique Laniel, Wenjun Yong, Richard A. Secco, John S. Tse, Serge Desgreniers, Paul A. Dube, Michael Shatruk, and Richard T. Oakley "Heat, Pressure and Light-Induced Interconversion of Bisdithiazolyl Radicals and Dimers", Journal of the American Chemical Society 2014 136 (22), 8050-8062 DOI: 10.1021/ja502753t.
- Jost, Marco; Born, David A.; Cracan, Valentin; Banerjee, Ruma; Drennan, Catherine L. (2015). "Structural Basis for Substrate Specificity in Adenosylcobalamin-dependent Isobutyryl-CoA Mutase and Related Acyl-CoA Mutases". Journal of Biological Chemistry. 290 (45): 26882–26898. doi:10.1074/jbc.M115.676890. PMC 4646380. PMID 26318610.
- Mendenhall, G. (1978). Long-lived free radicals. Science Progress (1933- ), 65(257), 1-18. Retrieved December 5, 2020, from http://www.jstor.org/stable/43420441
- Holger, S. P. Müller et al., J. Chem. Phys. 107, 8292 (1997); https://doi.org/10.1063/1.475030
- J. Org. Chem. 2006, 71, 3, 1209–1219 Publication Date:January 4, 2006 https://doi.org/10.1021/jo052363t


