Isotope-ratio mass spectrometry
Isotope-ratio mass spectrometry (IRMS) is a specialization of mass spectrometry, in which mass spectrometric methods are used to measure the relative abundance of isotopes in a given sample.[1][2]
 Magnetic sector mass spectrometer used in isotope ratio analysis, through thermal ionization. | |
| Acronym | IRMS |
|---|---|
| Classification | mass spectrometry |
This technique has two different applications in the earth and environmental sciences. The analysis of 'stable isotopes' is normally concerned with measuring isotopic variations arising from mass-dependent isotopic fractionation in natural systems. On the other hand, radiogenic isotope analysis[3] involves measuring the abundances of decay-products of natural radioactivity, and is used in most long-lived radiometric dating methods.
Introduction

The isotope-ratio mass spectrometer (IRMS) allows the precise measurement of mixtures of naturally occurring isotopes.[4] Most instruments used for precise determination of isotope ratios are of the magnetic sector type. This type of analyzer is superior to the quadrupole type in this field of research for two reasons. First, it can be set up for multiple-collector analysis, and second, it gives high-quality 'peak shapes'. Both of these considerations are important for isotope-ratio analysis at very high precision and accuracy.[3]
The sector-type instrument designed by Alfred Nier was such an advance in mass spectrometer design that this type of instrument is often called the 'Nier type'. In the most general terms the instrument operates by ionizing the sample of interest, accelerating it over a potential in the kilo-volt range, and separating the resulting stream of ions according to their mass-to-charge ratio (m/z). Beams with lighter ions bend at a smaller radius than beams with heavier ions. The current of each ion beam is then measured using a 'Faraday cup' or multiplier detector.
Many radiogenic isotope measurements are made by ionization of a solid source, whereas stable isotope measurements of light elements (e.g. H, C, O) are usually made in an instrument with a gas source. In a "multicollector" instrument, the ion collector typically has an array of Faraday cups, which allows the simultaneous detection of multiple isotopes.[5]
Gas source mass spectrometry
Measurement of natural variations in the abundances of stable isotopes of the same element is normally referred to as stable isotope analysis. This field is of interest because the differences in mass between different isotopes leads to isotope fractionation, causing measurable effects on the isotopic composition of samples, characteristic of their biological or physical history.
As a specific example, the hydrogen isotope deuterium (heavy hydrogen) is almost double the mass of the common hydrogen isotope. Water molecules containing the common hydrogen isotope (and the common oxygen isotope, mass 16) have a mass of 18. Water incorporating a deuterium atom has a mass of 19, over 5% heavier. The energy to vaporise the heavy water molecule is higher than that to vaporize the normal water so isotope fractionation occurs during the process of evaporation. Thus a sample of sea water will exhibit a quite detectable isotopic-ratio difference when compared to Antarctic snowfall.
Samples must be introduced to the mass spectrometer as pure gases, achieved through combustion, gas chromatographic feeds,[6] or chemical trapping. By comparing the detected isotopic ratios to a measured standard, an accurate determination of the isotopic make up of the sample is obtained. For example, carbon isotope ratios are measured relative to the international standard for C. The C standard is produced from a fossil belemnite found in the Peedee Formation, which is a limestone formed in the Cretaceous period in South Carolina, U.S.A. The fossil is referred to as VPDB (Vienna Pee Dee Belemnite) and has 13C:12C ratio of 0.0112372. Oxygen isotope ratios are measured relative the standard, V-SMOW (Vienna Standard Mean Ocean Water).

It is critical that the sample be processed before entering the mass spectrometer so that only a single chemical species enters at a given time. Generally, samples are combusted or pyrolyzed and the desired gas species (usually hydrogen (H2), nitrogen (N2), carbon dioxide (CO2), or sulfur dioxide (SO2)) is purified by means of traps, filters, catalysts and/or chromatography.
The two most common types of IRMS instruments are continuous flow[7] and dual inlet. In dual inlet IRMS, purified gas obtained from a sample is alternated rapidly with a standard gas (of known isotopic composition) by means of a system of valves, so that a number of comparison measurements are made of both gases. In continuous flow IRMS, sample preparation occurs immediately before introduction to the IRMS, and the purified gas produced from the sample is measured just once. The standard gas may be measured before and after the sample or after a series of sample measurements. While continuous-flow IRMS instruments can achieve higher sample throughput and are more convenient to use than dual inlet instruments, the yielded data is of approximately 10-fold lower precision.
Static gas mass spectrometry
A static gas mass spectrometer is one in which a gaseous sample for analysis is fed into the source of the instrument and then left in the source without further supply or pumping throughout the analysis. This method can be used for 'stable isotope' analysis of light gases (as above), but it is particularly used in the isotopic analysis of noble gases (rare or inert gases) for radiometric dating or isotope geochemistry. Important examples are argon–argon dating and helium isotope analysis.
Thermal ionization mass spectrometry
Several of the isotope systems involved in radiometric dating depend on IRMS using thermal ionization of a solid sample loaded into the source of the mass spectrometer (hence thermal ionization mass spectrometry, TIMS). These methods include rubidium–strontium dating, uranium–lead dating, lead–lead dating and samarium–neodymium dating.
When these isotope ratios are measured by TIMS, mass-dependent fractionation occurs as species are emitted by the hot filament. Fractionation occurs due to the excitation of the sample and therefore must be corrected for accurate measurement of the isotope ratio.[8]
There are several advantages of the TIMS method. It has a simple design, is less expensive than other mass spectrometers, and produces stable ion emissions. It requires a stable power supply, and is suitable for species with a low ionization potential, such as Strontium (Sr), and Lead (Pb).
The disadvantages of this method stem from the maximum temperature achieved in thermal ionization. The hot filament reaches a temperature of less than 2500 degrees Celsius, leading to the inability to create atomic ions of species with a high ionization potential, such as Osmium (Os), and Tungsten (Hf-W). Although the TIMS method can create molecular ions instead in this case, species with high ionization potential can be analyzed more effectively with MC-ICP-MS.
Secondary-ion mass spectrometry
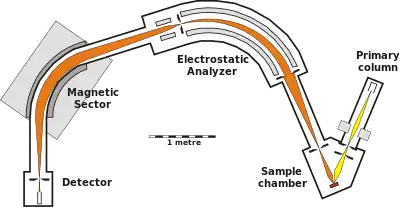
An alternative approach used to measure the relative abundance of radiogenic isotopes when working with a solid surface is secondary-ion mass spectrometry (SIMS). This type of ion-microprobe analysis normally works by focusing a primary (oxygen) ion beam on a sample in order to generate a series of secondary positive ions that can be focused and measured based on their mass/charge ratios.
SIMS is a common method used in U-Pb analysis, as the primary ion beam is used to bombard the surface of a single zircon grain in order to yield a secondary beam of Pb ions. The Pb ions are analyzed using a double focusing mass spectrometer that comprises both an electrostatic and magnetic analyzer. This assembly allows the secondary ions to be focused based on their kinetic energy and mass-charge ratio in order to be accurately collected using a series of Faraday cups.[10]
A major issue that arises in SIMS analysis is the generation of isobaric interference between sputtered molecular ions and the ions of interest. This issue occurs with U–Pb dating as Pb ions have essentially the same mass as HfO2+.[11] In order to overcome this problem, a sensitive high-resolution ion microprobe (SHRIMP) can be used. A SHRIMP is a double-focusing mass spectrometer that allows for a large spatial separation between different ion masses based on its relatively large size. For U-Pb analysis, the SHRIMP allows for the separation of Pb from other interfering molecular ions, such as HfO2+.
Multiple collector inductively coupled plasma mass spectrometry
An MC-ICP-MS instrument is a multiple collector mass spectrometer with a plasma source. MC-ICP-MS was developed to improve the precision achievable by ICP-MS during isotope-ratio measurements. Conventional ICP-MS analysis uses a quadrupole analyser, which only allows single-collector analysis. Due to the inherent instability of the plasma, this limits the precision of ICP-MS with a quadrupole analyzer to around 1%, which is insufficient for most radiogenic isotope systems.
Isotope-ratio analysis for radiometric dating has normally been determined by TIMS. However, some systems (e.g. Hf-W and Lu-Hf) are difficult or impossible to analyse by TIMS, due to the high ionization potential of the elements involved. Therefore, these methods can now be analysed using MC-ICP-MS.
The Ar-ICP produces an ion-beam with a large inherent kinetic energy distribution, which makes the design of the mass-spectrometer somewhat more complex than it is the case for conventional TIMS instruments. First, different from Quadrupole ICP-MS systems, magnetic sector instruments have to operate with a higher acceleration potential (several 1000 V) in order to minimize the energy distribution of the ion beam. Modern instruments operate at 6-10kV. The radius of deflection of an ion within a magnetic field depends on the kinetic energy and the mass/charge ratio of the ion (strictly, the magnet is a momentum analyzer not just a mass analyzer). Because of the large energy distribution, ions with similar mass/charge ratio can have very different kinetic energies and will thus experience different deflection for the same magnetic field. In practical terms one would see that ions with the same mass/charge ratio focus at different points in space. However, in a mass-spectrometer one wants ions with the same mass/charge ratio to focus at the same point, e.g. where the detector is located. In order to overcome these limitations, commercial MC-ICP-MS are double-focusing instruments. In a double-focusing mass-spectrometer ions are focused due to kinetic energy by the ESA (electro-static-analyzer) and kinetic energy + mass/charge (momentum) in the magnetic field. Magnet and ESA are carefully chosen to match the energy focusing properties of one another and are arranged so that the direction of energy focusing is in opposite directions. To simplify, two components have an energy focus term, when arranged properly, the energy term cancels out and ions with the same mass/charge ratio focus at the same point in space. It is important to note, double-focusing does not reduce the kinetic energy distribution and different kinetic energies are not filtered or homogenized. Double-focusing works for single as well as multi-collector instruments. In single collector instruments ESA and magnet can be arranged in either forward geometry (first ESA then magnet) or reversed geometry (magnet first then ESA), as only point-to-point focusing is required. In multi-collector instruments, only forward geometry (ESA then magnet) is possible due to the array of detectors and the requirements of a focal plane rather than a focal point.
Accelerator mass spectrometry
 Accelerator mass spectrometer at Lawrence Livermore National Laboratory | |
| Acronym | AMS |
|---|---|
| Classification | Mass spectrometry |
| Analytes | Organic molecules Biomolecules |
| Other techniques | |
| Related | Particle accelerator |
For isotopes occurring at extremely low levels, accelerator mass spectrometry (AMS) can be used. For example, the decay rate of the radioisotope 14C is widely used to date organic materials, but this approach was once limited to relatively large samples no more than a few thousand years old. AMS extended the range of 14C dating to about 60,000 years BP, and is about 106 times more sensitive than conventional IRMS.
AMS works by accelerating negative ions through a large (mega-volt) potential, followed by charge exchange and acceleration back to ground. During charge exchange, interfering species can be effectively removed. In addition, the high energy of the beam allows the use of energy-loss detectors, that can distinguish between species with the same mass/charge ratio. Together, these processes allow the analysis of extreme isotope ratios above 1012.
Moving wire IRMS
Moving wire IRMS is useful for analyzing carbon-13 ratios of compounds in a solution, such as after purification by liquid chromatography. The solution (or outflow from the chromatography) is dried onto a nickel or stainless steel wire. After the residue is deposited on the wire, it enters a furnace where the sample is converted to CO2 and water by combustion. The gas stream finally enters a capillary, is dried, ionized, and analyzed.[12] This process allows a mixture of compounds to be purified and analyzed continuously, which can decrease the analysis time by a factor of four.[12] Moving wire IRMS is quite sensitive, and samples containing as little as 1 nanomole of carbon can yield precise (within 1‰) results.[13]
References
- Paul D, Skrzypek G, Fórizs I (2007). "Normalization of measured stable isotopic compositions to isotope reference scales - a review". Rapid Commun. Mass Spectrom. 21 (18): 3006–14. Bibcode:2007RCMS...21.3006P. doi:10.1002/rcm.3185. PMID 17705258.
- Stellaard F, Elzinga H (2005). "Analytical techniques in biomedical stable isotope applications: (isotope ratio) mass spectrometry or infrared spectrometry?". Isotopes in Environmental and Health Studies. 41 (4): 345–61. doi:10.1080/10256010500384333. PMID 16543190.
- Dickin, A.P. (2005). Radiogenic Isotope Geology. Cambridge University Press. Archived from the original on 2014-03-27. Retrieved 2008-10-09.
- Townsend, A., ed. (1995). Encyclopaedia of Analytical Science Encyclopaedia of Analytical Science. London: Academic Press Limited.
- C. B. Bouthitt; K. Garnett. "The Evolution of the Multicollector in Isotope Ratio Mass Spectromety". Proceedings of the 18th AMZSMS Conference: THO–07.
- Meier-Augenstein, W. (1999). "Applied gas chromatography coupled to isotope ratio mass spectrometry". J. Chromatogr. A. 842 (1–2): 351–371. doi:10.1016/S0021-9673(98)01057-7. PMID 10377971.
- Brenna JT, Corso TN, Tobias HJ, Caimi RJ (1997). "High-precision continuous-flow isotope ratio mass spectrometry". Mass Spectrometry Reviews. 16 (5): 227–58. Bibcode:1997MSRv...16..227B. doi:10.1002/(SICI)1098-2787(1997)16:5<227::AID-MAS1>3.0.CO;2-J. PMID 9538528.
- Dickin, A.P., 2005. Radiogenic Isotope Geology 2nd ed. (Cambridge: Cambridge University Press), pp. 21-22.
- Williams, I.S. (1998), "U-Th-Pb geochronology by ion microprobe", In: McKibben, M.A.; Shanks III, W.C.; Ridley, W.I.; (Editors), "Applications of microanalytical techniques to understanding mineralizing processes", Reviews in Economic Geology Special Publication 7: 1–35
- Dickin, A. P. (2005). Radiogenic Isotope Geology 2nd ed. Cambridge University Press.
- Hinton, R.W. and Long, J.V.P. (1979). High-resolution ion microprobe measurement of lead isotopes: variations within single zircons from Lac Seul, Northwestern Ontario. Earth Planet. Sci. lett. 45, 309-325.,
- Caimi, R. J.; Brenna, J. T. (1996). "Direct analysis of carbon isotope variability in albumins by liquid flow-injection isotope ratio mass spectrometry". J. Am. Soc. Mass Spectrom. 7 (6): 605–610. doi:10.1016/1044-0305(96)00010-4. PMID 24203433.
- Sessions, A.L.; Sylva, S.P.; Hayes, J.M. (2005). "Moving-wire device for carbon isotopic analyses of nanogram quantities of nonvolatile organic carbon". Analytical Chemistry. 77 (20): 6519–6527. doi:10.1021/ac051251z. PMID 16223235.
Bibliography
- Goetz, A.; Platzner, I. T. (Itzhak Thomas); Habfast, K.; Walder, A. J. (1997). Modern isotope ratio mass spectrometry. London: J. Wiley. ISBN 978-0-471-97416-1. OCLC 36461690.
- Yamasaki, Shin-ichi; Boutton, Thomas W. (1996). Mass spectrometry of soils. New York: M. Dekker. ISBN 978-0-8247-9699-0. OCLC 34473560.
| Ion source | |
|---|---|
| Mass analyzer | |
| Detector | |
| MS combination | |
| Fragmentation | |
| |