Lujan–Fryns syndrome
Lujan–Fryns syndrome (LFS) is an X-linked genetic disorder that causes mild to moderate intellectual disability and features described as Marfanoid habitus, referring to a group of physical characteristics similar to those found in Marfan syndrome.[4][5] These features include a tall, thin stature and long, slender limbs.[5] LFS is also associated with psychopathology and behavioral abnormalities, and it exhibits a number of malformations affecting the brain and heart.[6][7][8] The disorder is inherited in an X-linked dominant manner, and is attributed to a missense mutation in the MED12 gene.[3] There is currently no treatment or therapy for the underlying MED12 malfunction, and the exact cause of the disorder remains unclear.[9]
| Lujan–Fryns syndrome | |
|---|---|
| Other names | X-linked mental retardation with Marfanoid habitus, Lujan syndrome[1][2][3] |
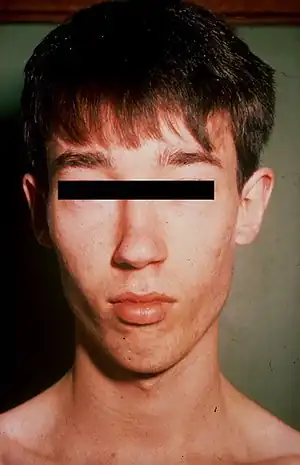 | |
| Lujan–Fryns syndrome in a young adult male, with features that include a long, narrow face and recessed chin. | |
| Specialty | Medical genetics |
Signs and symptoms
Intellectual disability in LFS usually ranges from mild to moderate, but severe cases have also been reported.[10][11] A relatively common brain anomaly seen with LFS is agenesis of the corpus callosum, an error of embryonic development in which the corpus callosum (a structure of the mammalian brain composed of nerves that allows communication between the left and right cerebral hemispheres) is not present.[7][12] Among a number of adverse neurological effects sometimes found with an absence of the corpus callosum, intellectual disability has been shown to occur at a rate of approximately 73 percent.[12] A correlation between agenesis of the corpus callosum and intellectual disability in LFS, however, has not been suggested.[13]
Psychiatric
Psychopathology and related behavioral abnormalities are typically seen in LFS, and they may be considered in the diagnosis of the disorder.[7] The most common of these in LFS is an autism-like spectrum disorder, and LFS is considered one of a number of genetic disorders associated with autism.[7][14] Additional alterations of psychopathology with behavioral manifestations that have been observed in LFS include: psychotic behavior,[15] schizophrenia,[16] hyperactivity and attention-deficit hyperactivity disorder,[13][17] aggression,[17] oppositional defiant disorder,[13][18] obsessive compulsive disorder,[13] extreme shyness,[17] learning disability,[13] cognitive impairment,[13] short-term memory deficit,[13] low frustration tolerance,[13] social dysfunction,[13] lack of impulse control,[13] eating disorder and associated malnutrition, attributed to psychogenic loss of appetite;[6] and pyromania.[7][13][18]
While psychiatric conditions like these are to be expected with LFS, there have also been cases of the disorder with some preservation of mental and behavioral abilities, such as problem solving, reasoning and normal intelligence.[19]
The psychopathology of LFS usually exhibits schizophrenia.[16] When schizophrenia is diagnosed in an individual known to be affected by intellectual disability, LFS may be considered in the differential diagnosis of schizophrenia, with confirmation of cause through appropriate psychiatric and genetic evaluation methods.[16]
Marfanoid habitus
LFS is clinically distinguished from other X-linked forms of intellectual disability by the accompanying presence of marfanoid habitus.[10] Marfanoid habitus describes a group of physical features common to Marfan syndrome.[5] Including Marfan syndrome and LFS, marfanoid features of this type have also been observed with several other disorders, one of which is multiple endocrine neoplasia type 2.[20]
In LFS, specific features identified as marfanoid include: a long, narrow face;[5][9] tall, thin stature;[3][9] long, slender limbs, fingers and toes (not unlike arachnodactyly)[3][21][22] with joint hyperextensibility,[17] shortened halluces (the big toes) and long second toes.[9]
The diagnosis of marfanoid habitus in LFS is often delayed because many of the physical features and characteristics associated with it are usually not evident until adolescence.[2]
Head and face
Craniofacial and other features of LFS include: maxillary hypoplasia (underdevelopment of the upper jaw bone),[9] a small mandible (lower jaw bone) and receding chin,[3][17] a high-arched palate (the roof of the mouth), with crowding and misalignment of the upper teeth;[5][7] macrocephaly (enlarged skull) with a prominent forehead,[3][9] hypernasal speech (voice),[5][7] a long nose with a high, narrow nasal bridge;[9] a deep, short philtrum (the indentation in the upper lip, beneath the nose),[9] low-set ears with some apparent retroversion,[9] hypotonia (decreased muscle tone),[3] pectus excavatum (a malformity of the chest),[9] slightly enlarged to normal testicular size in males,[9][17] and seizures.[9]
Hypernasal speech, or "hypernasality", is primarily the result of velopharyngeal insufficiency, a sometimes congenital aberration in which the velopharyngeal sphincter allows too much air into the nasal cavity during speech.[23][24] In LFS, hypernasality may also be caused by failure of the soft palate and uvula to reach the back wall of the pharynx (the interior cavity of the throat where swallowing generally occurs) during speech, a condition that can be associated with a submucosal cleft palate.[13][25]
Heart
A number of features involving the heart have been noted in several LFS cases, the most significant being dilation of the aortic root, a section of the ascending aorta.[8] Aortic root dilation (enlargement) is associated with a greatly increased risk of dissection of the aortic wall, resulting in aortic aneurysm.[26] As this presents a possible life-threatening consequence of LFS, routine cardiac evaluation methods such as echocardiogram are implemented when the disorder is first diagnosed, along with MRI scans of the brain to screen for suspected agenesis of the corpus callosum.[7] Additional effects on the heart that have been reported with LFS are ventricular and atrial septal defect.[8][17]
Cause
A missense mutation in the MED12 gene, located on the human X chromosome, has been established as the cause of LFS.[3][27] Missense mutations are genetic point mutations in which a single nucleotide in the genetic sequence is exchanged with another one. This leads to an erroneously substitution of a particular amino acid in the protein sequence during translation. The missense mutation in the MED12 gene, that causes LFS, is identified as p.N1007S.[3] This indicates that the amino acid asparagine, normally located at position 1007 along the MED12 sequence, has been mistakenly replaced by serine.[27] This mutation in MED12 causes incorrect expression and activity of the protein it encodes, resulting in the disorder.[3][9]
Pathophysiology
MED12, or mediator of RNA polymerase II transcription, subunit 12 homolog of S. cerevisiae, is one of several subunits in the mammalian mediator complex, which regulates RNA polymerase II during mRNA transcription.[28][29]
The Mediator complex is required for polymerase II transcription and acts as a bridge between the polymerase II enzyme and different gene-specific transcription factors. Mediator can contain up to 30 subunits, but some of the subunits are only required for regulation of transcription in particular tissues or cells.[30] Currently, the exact mechanism by which dysfunction of MED12 results in LFS and its associated neuropsychopathic and physical characteristics is unclear. Marfanoid habitus, a highly arched palate and several other features of LFS can be found with Marfan syndrome, a connective tissue disorder.[4] The finding of aortic root dilation in both disorders suggests that a mutation in an unspecified connective tissue regulating gene may contribute to the etiology of LFS.[1][5][8][13]
A number of interesting experimental results have been obtained by studying MED12 mutations in the zebrafish, an animal model representing vertebrates.[31][32][33] In zebrafish, a mutation in MED12 was found to be responsible for the mutant motionless (mot). Zebrafish with the mot mutation have neuronal and cardiovascular defects, although not all types of neurons are affected. Introduction of human MED12 mRNA into the zebrafish restores normal development.[34] MED12 is also a critical coactivator for the gene SOX9, which is involved in the developmental regulation of neurons, cartilage and bone. In the zebrafish, MED12 defects cause maldevelopment of vertebrate embryonic structures such as the neural crest, which would alter function of the autonomic and peripheral nervous systems; and they also cause malformations of cell types serving as precursors to cartilage and bone, such as osteocytes.[34][35][36] Some features found in LFS, like agenesis of the corpus callosum and cartilage-related craniofacial anomalies, are similar to defects found in zebrafish with MED12 and associated mutations.[3]
Genetics
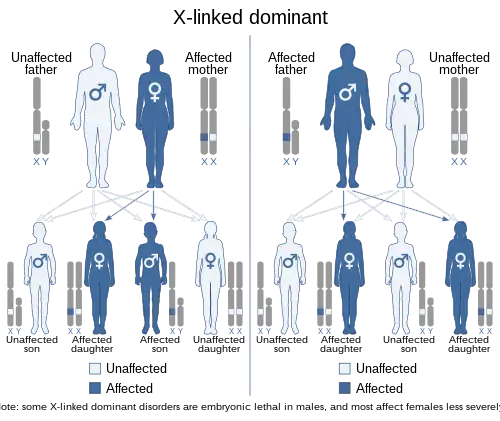
Lujan–Fryns syndrome is inherited in an X-linked dominant manner.[9][13][37] This means the defective gene responsible for the disorder (MED12) is located on the X chromosome, and only one copy of the defective gene is sufficient to cause the disorder when inherited from a parent who has the disorder. Males are normally hemizygous for the X chromosome, having only one copy. As a result, X-linked dominant disorders usually show higher expressivity in males than females. This phenomenon is thought to occur with LFS.[13][37]
As the X chromosome is one of the sex chromosomes (the other being the Y chromosome), X-linked inheritance is determined by the gender of the parent carrying a specific gene and can often seem complex. This is because, typically, females have two copies of the X-chromosome, while males have only one copy. The difference between dominant and recessive inheritance patterns also plays a role in determining the chances of a child inheriting an X-linked disorder from their parentage.
In LFS, X-linked dominant inheritance was suspected, as boy and girl siblings in one family both exhibited the disorder.[13][37] A scenario such as this would also be possible with X-linked recessive inheritance, but in this particular case report, the girl was believed to be a manifesting heterozygote[13][37] carrying one copy of the mutated gene.
Sporadic cases of LFS, where the disorder is present in an individual with no prior family history of it, have also been reported in a small number of affected males.[13][15][38]
Similarities to other genetic diseases
An individual exhibiting intellectual disability and other symptoms similar to LFS was found to have a terminal deletion of the subtelomeric region in the short arm of chromosome 5.[25] Deletion of this area of chromosome 5 is associated with intellectual disability, psychotic behavior, autism, macrocephaly and hypernasal-like speech, as well as the disorder Cri du chat syndrome.[25][39] Fryns (2006) suggests a detailed examination of chromosome 5 with FISH should be performed as part of the differential diagnosis of LFS.[9]
Mutations in the UPF3B gene, also found on the X chromosome, are another cause of X-linked intellectual disability.[40] UPF3B is part of the nonsense-mediated mRNA decay (NMD) complex, which performs mRNA surveillance, detecting mRNA sequences that have been erroneously truncated (shortened) by the presence of nonsense mutations.[41] Mutations in UPF3B alter and prevent normal function of the NMD pathway, resulting in translation and expression of truncated mRNA sequences into malfunctioning proteins that can be associated with developmental errors and intellectual disability.[41][42] Individuals from two families diagnosed with LFS and one family with FGS were found to have mutations in UPF3B, confirming that the clinical presentations of the different mutations can overlap.[42]
Diagnosis
Although LFS is usually suspected when intellectual disability and marfanoid habitus are observed together in a patient, the diagnosis of LFS can be confirmed by the presence of the p.N1007S missense mutation in the MED12 gene.[3][9][10]
Differential diagnosis
In the differential diagnosis of LFS, another disorder that exhibits some features and symptoms of LFS and is also associated with a missense mutation of MED12 is Opitz-Kaveggia syndrome (FGS).[3][43] Common features shared by both LFS and FGS include X-linked intellectual disability, hyperactivity, macrocephaly, corpus callosum agenesis and hypotonia.[3] Notable features of FGS that have not been reported with LFS include excessive talkativeness, consistent strength in socialization skills, imperforate anus (occlusion of the anus) and ocular hypertelorism (extremely wide-set eyes).[44][45]
Whereas LFS is associated with missense mutation p.N1007S, FGS is associated with missense mutation p.R961W.[3][46] As both disorders originate from an identical type of mutation in the same gene, while exhibiting similar, yet distinct characteristics; LFS and FGS are considered to be allelic.[3][9][13][43] In the context of MED12, this suggests that the phenotype of each disorder is related to the way in which their respective mutations alter the MED12 sequence and its function.[3][27][43]
Treatment
While there is no specific treatment for the underlying genetic cause of LFS, corrective procedures, preventive intervention measures, and therapies may be considered in the treatment and management of the many craniofacial, orthopedic, and psychiatric problems associated with the disorder. More pressing issues such as cardiac involvement or epileptic seizures should be routinely examined and monitored. Close attention and specialized follow-up care, including neuro-psychological evaluation methods and therapies, and special education, should be given to diagnose and prevent psychiatric disorders and related behavioral problems such as psychosis and outbursts of aggression.[9]
Epidemiology
Lujan–Fryns syndrome is a rare X-linked dominant syndrome and is more common in males than females. Its prevalence within the general population has not yet been determined.[9]
History
Lujan–Fryns syndrome is named after physicians J. Enrique Lujan and Jean-Pierre Fryns.[21] The initial observation of suspected X-linked intellectual disability with Marfanoid features and craniofacial effects such as a high-arched palate was described by Lujan et al. in 1984.[17] In the report, four affected male members of a large kindred (consanguinous family) were noted.[3][13][17] Additional investigations of combined X-linked intellectual disability and Marfanoid habitus in other families, including two brothers, were reported by Fryns et al., beginning in 1987.[5] The disorder soon became known as Lujan–Fryns syndrome.[37]
References
- Lacombe, D.; Bonneau, D.; Verloes, A.; Couet, D.; Koulischer, L.; Battin, J. (1993). "Lujan-Fryns syndrome (X-linked mental retardation with marfanoid habitus): report of three cases and review". Genetic Counseling (Geneva, Switzerland). 4 (3): 193–198. ISSN 1015-8146. PMID 8267926.
- Fryns, J. P.; Van Den Berghe, H. (1991). "X-linked mental retardation with Marfanoid habitus: a changing phenotype with age?". Genetic Counseling (Geneva, Switzerland). 2 (4): 241–244. ISSN 1015-8146. PMID 1799424.
- Schwartz, C. E.; Tarpey, P. S.; Lubs, H. A.; Verloes, A.; May, M. M.; Risheg, H.; Friez, M. J.; Futreal, P. A.; Edkins, S.; Teague, J.; Briault, S.; Skinner, C.; Bauer-Carlin, A.; Simensen, R. J.; Joseph, S. M.; Jones, J. R.; Gecz, J.; Stratton, M. R.; Raymond, F. L.; Stevenson, R. E. (July 2007). "The original Lujan syndrome family has a novel missense mutation (p.N1007S) in the MED12 gene". Journal of Medical Genetics. 44 (7): 472–477. doi:10.1136/jmg.2006.048637. ISSN 0022-2593. PMC 2597996. PMID 17369503.
- Online Mendelian Inheritance in Man (OMIM): 154700
- Fryns, J. P.; Buttiens, M.; Opitz, J. M.; Reynolds, J. F. (Oct 1987). "X-linked mental retardation with marfanoid habitus". American Journal of Medical Genetics. 28 (2): 267–274. doi:10.1002/ajmg.1320280202. ISSN 0148-7299. PMID 3322000.
- Alonso, P.; Pintos, G.; Almazan, F.; Hernández, L.; Loran, E.; Menchon, J. M.; Vallejo, J. (July 2006). "Eating disorder in a patient with phenotypical features of Lujan-Fryns syndrome". Clinical Dysmorphology. 15 (3): 181–184. doi:10.1097/01.mcd.0000220610.24908.a4. ISSN 0962-8827. PMID 16760741. S2CID 7415391.
- Lerma‐Carrillo, I.; Molina, J. D.; Cuevas-Duran, T.; Julve-Correcher, C.; Espejo-Saavedra, J. M.; Andrade-Rosa, C.; Lopez-Muñoz, F. (December 2006). "Psychopathology in the Lujan-Fryns syndrome: report of two patients and review". American Journal of Medical Genetics Part A. 140 (24): 2807–2811. doi:10.1002/ajmg.a.31503. ISSN 1552-4825. PMID 17036352. S2CID 22491132.
- Wittine, L. M.; Josephson, K. D.; Williams, M. S. (Oct 1999). "Aortic root dilation in apparent Lujan-Fryns syndrome". American Journal of Medical Genetics. 86 (5): 405–409. doi:10.1002/(SICI)1096-8628(19991029)86:5<405::AID-AJMG2>3.0.CO;2-1. ISSN 0148-7299. PMID 10508979.
- Buggenhout, G. V.; Fryns, J. -P. (July 2006). "Lujan-Fryns syndrome (mental retardation, X-linked, marfanoid habitus)". Orphanet Journal of Rare Diseases (Free full text). 1: 26. doi:10.1186/1750-1172-1-26. PMC 1538574. PMID 16831221.
- Fryns, J. P.; Buttiens, M.; Van Den Berghe, H. (Jan 1988). "Chromosome X-linked mental retardation and marfanoid syndrome". Journal de Génétique Humaine. 36 (1–2): 123–128. ISSN 0021-7743. PMID 3379374.
- Mégarbané A, C. C.; Chammas, C. (1997). "Severe mental retardation with marfanoid habitus in a young Lebanese male. A diagnostic challenge". Genetic Counseling (Geneva, Switzerland). 8 (3): 195–200. ISSN 1015-8146. PMID 9327261.
- Jeret, J. S.; Serur, D.; Wisniewski, K. E.; Lubin, R. A. (1987). "Clinicopathological findings associated with agenesis of the corpus callosum". Brain & Development. 9 (3): 255–264. doi:10.1016/s0387-7604(87)80042-6. ISSN 0387-7604. PMID 3310713. S2CID 4761497.
- Online Mendelian Inheritance in Man (OMIM): 309520
- Artigas-Pallarés, J.; Gabau-Vila, E.; Guitart-Feliubadaló, M. (Jan 2005). "Syndromic autism: II. Genetic syndromes associated with autism". Revista de Neurología. 40 (Suppl 1): S151–S162. doi:10.33588/rn.40S01.2005073. ISSN 0210-0010. PMID 15736079.
- Lalatta, F.; Livini, E.; Selicorni, A.; Briscioli, V.; Vita, A.; Lugo, F.; Zollino, M.; Gurrieri, F.; Neri, G. (Feb 1991). "X-linked mental retardation with marfanoid habitus: first report of four Italian patients". American Journal of Medical Genetics. 38 (2–3): 228–232. doi:10.1002/ajmg.1320380211. ISSN 0148-7299. PMID 2018063.
- De Hert, M.; Steemans, D.; Theys, P.; Fryns, J. P.; Peuskens, J. (Apr 1996). "Lujan-Fryns syndrome in the differential diagnosis of schizophrenia". American Journal of Medical Genetics. 67 (2): 212–213. doi:10.1002/(SICI)1096-8628(19960409)67:2<212::AID-AJMG13>3.0.CO;2-M. PMID 8723050.
- Lujan, J. E.; Carlin, M. E.; Lubs, H. A.; Opitz, J. M. (Jan 1984). "A form of X-linked mental retardation with marfanoid habitus". American Journal of Medical Genetics. 17 (1): 311–322. doi:10.1002/ajmg.1320170124. ISSN 0148-7299. PMID 6711603.
- Williams, M. S. (Dec 2006). "Neuropsychological evaluation in Lujan-Fryns syndrome: commentary and clinical report". American Journal of Medical Genetics Part A. 140 (24): 2812–2815. doi:10.1002/ajmg.a.31501. ISSN 1552-4825. PMID 17103446. S2CID 29096814.
- Donders, J.; Toriello, H.; Van Doornik, S. (Jan 2002). "Preserved neurobehavioral abilities in Lujan-Fryns syndrome". American Journal of Medical Genetics. 107 (3): 243–246. doi:10.1002/ajmg.10144. ISSN 0148-7299. PMID 11807907.
- Prabhu, M.; Khouzam, R. N.; Insel, J. (Nov 2004). "Multiple endocrine neoplasia type 2 syndrome presenting with bowel obstruction caused by intestinal neuroma: case report". Southern Medical Journal. 97 (11): 1130–1132. doi:10.1097/01.SMJ.0000140873.29381.12. ISSN 0038-4348. PMID 15586612. S2CID 27428744.
- synd/3838 at Who Named It?
- Buntinx, I. M.; Willems, P. J.; Spitaels, S. E.; Van Reempst, P. J.; De Paepe, A. M.; Dumon, J. E. (April 1991). "Neonatal Marfan syndrome with congenital arachnodactyly, flexion contractures, and severe cardiac valve insufficiency". Journal of Medical Genetics. 28 (4): 267–273. doi:10.1136/jmg.28.4.267. ISSN 0022-2593. PMC 1016831. PMID 1856834.
- Willging, J. P. (Oct 1999). "Velopharyngeal insufficiency". International Journal of Pediatric Otorhinolaryngology. 49 (Suppl 1): S307–S309. doi:10.1016/S0165-5876(99)00182-2. ISSN 0165-5876. PMID 10577827.
- Warren, D. W.; Dalston, R. M.; Mayo, R. (Jul 1994). "Hypernasality and velopharyngeal impairment". The Cleft Palate-Craniofacial Journal. 31 (4): 257–262. doi:10.1597/1545-1569(1994)031<0257:HAVI>2.3.CO;2. ISSN 1055-6656. PMID 7918520.
- Stathopulu, E.; Ogilvie, C. M.; Flinter, F. A. (June 2003). "Terminal deletion of chromosome 5p in a patient with phenotypical features of Lujan-Fryns syndrome". American Journal of Medical Genetics Part A. 119A (3): 363–366. doi:10.1002/ajmg.a.10268. ISSN 1552-4825. PMID 12784307. S2CID 45722356.
- Gambarin, F.; Favalli, V.; Serio, A.; Regazzi, M.; Pasotti, M.; Klersy, C.; Dore, R.; Mannarino, S.; Viganò, M.; Odero, A.; Amato, S.; Tavazzi, L.; Arbustini, E. (April 2009). "Rationale and design of a trial evaluating the effects of losartan vs. Nebivolol vs. The association of both on the progression of aortic root dilation in Marfan syndrome with FBN1 gene mutations". Journal of Cardiovascular Medicine (Hagerstown, Md.). 10 (4): 354–362. doi:10.2459/JCM.0b013e3283232a45. ISSN 1558-2027. PMID 19430350. S2CID 29419873.
- Online Mendelian Inheritance in Man (OMIM): 300188
- Biddick, R.; Young, E. (Sep 2005). "Yeast mediator and its role in transcriptional regulation". Comptes Rendus Biologies. 328 (9): 773–782. doi:10.1016/j.crvi.2005.03.004. ISSN 1631-0691. PMID 16168358.
- Sims, R. J. 3rd; Mandal, S. S.; Reinberg, D. (June 2004). "Recent highlights of RNA-polymerase-II-mediated transcription". Current Opinion in Cell Biology. 16 (3): 263–271. doi:10.1016/j.ceb.2004.04.004. ISSN 0955-0674. PMID 15145350.
- Malik, S.; Roeder, R. G. (Jun 2000). "Transcriptional regulation through Mediator-like coactivators in yeast and metazoan cells". Trends in Biochemical Sciences. 25 (6): 277–283. doi:10.1016/S0968-0004(00)01596-6. ISSN 0968-0004. PMID 10838567.
- Chakraborty C, H. C.; Hsu, C. H.; Wen, Z. H.; Lin, C. S.; Agoramoorthy, G. (Feb 2009). "Zebrafish: a complete animal model for in vivo drug discovery and development". Current Drug Metabolism. 10 (2): 116–124. doi:10.2174/138920009787522197. ISSN 1389-2002. PMID 19275547.
- Kari, G.; Rodeck, U.; Dicker, A. P. (July 2007). "Zebrafish: an emerging model system for human disease and drug discovery". Clinical Pharmacology and Therapeutics. 82 (1): 70–80. doi:10.1038/sj.clpt.6100223. ISSN 0009-9236. PMID 17495877. S2CID 41443542.
- McGonnell, I. M.; Fowkes, R. C. (June 2006). "Fishing for gene function--endocrine modelling in the zebrafish" (Free full text). The Journal of Endocrinology. 189 (3): 425–439. doi:10.1677/joe.1.06683. ISSN 0022-0795. PMID 16731775.
- Wang, X.; Yang, N.; Uno, E.; Roeder, R. G.; Guo, S. (November 2006). "A subunit of the mediator complex regulates vertebrate neuronal development". Proceedings of the National Academy of Sciences of the United States of America (Free full text). 103 (46): 17284–17289. Bibcode:2006PNAS..10317284W. doi:10.1073/pnas.0605414103. ISSN 0027-8424. PMC 1859923. PMID 17088561.
- Rau, M. J.; Fischer, S.; Neumann, C. J. (Aug 2006). "Zebrafish Trap230/Med12 is required as a coactivator for Sox9-dependent neural crest, cartilage and ear development". Developmental Biology. 296 (1): 83–93. doi:10.1016/j.ydbio.2006.04.437. ISSN 0012-1606. PMID 16712834.
- Hong, S. -K.; Haldin, C. E.; Lawson, N. D.; Weinstein, B. M.; Dawid, I. B.; Hukriede, N. A. (December 2005). "The zebrafish kohtalo/trap230 gene is required for the development of the brain, neural crest, and pronephric kidney". Proceedings of the National Academy of Sciences of the United States of America. 102 (51): 18473–18478. Bibcode:2005PNAS..10218473H. doi:10.1073/pnas.0509457102. ISSN 0027-8424. PMC 1311743. PMID 16344459.
- Gurrieri, F.; Neri, G. (Feb 1991). "A girl with the Lujan-Fryns syndrome". American Journal of Medical Genetics. 38 (2–3): 290–291. doi:10.1002/ajmg.1320380225. ISSN 0148-7299. PMID 2018074.
- Fryns, J. P. (Feb 1991). "X-linked mental retardation with marfanoid habitus". American Journal of Medical Genetics. 38 (2–3): 233. doi:10.1002/ajmg.1320380212. ISSN 0148-7299. PMID 2018064.
- Fang, J. S.; Lee, K. F.; Huang, C. T.; Syu, C. L.; Yang, K. J.; Wang, L. H.; Liao, D. L.; Chen, C. H. (Jun 2008). "Cytogenetic and molecular characterization of a three-generation family with chromosome 5p terminal deletion". Clinical Genetics. 73 (6): 585–590. doi:10.1111/j.1399-0004.2008.00995.x. ISSN 0009-9163. PMID 18400035. S2CID 6209765.
- Online Mendelian Inheritance in Man (OMIM): 300298
- Chang, Y. F.; Imam, J. S.; Wilkinson, M. F. (2007). "The nonsense-mediated decay RNA surveillance pathway". Annual Review of Biochemistry. 76: 51–74. doi:10.1146/annurev.biochem.76.050106.093909. ISSN 0066-4154. PMID 17352659.
- Tarpey, P. S.; Raymond, F. L.; Nguyen, L. S.; Rodriguez, J.; Hackett, A.; Vandeleur, L.; Smith, R.; Shoubridge, C.; Edkins, S.; Stevens, C.; O'Meara, S.; Tofts, C.; Barthorpe, S.; Buck, G.; Cole, J.; Halliday, K.; Hills, K.; Jones, D.; Mironenko, T.; Perry, J.; Varian, J.; West, S.; Widaa, S.; Teague, J.; Dicks, E.; Butler, A.; Menzies, A.; Richardson, D.; Jenkinson, A.; Shepherd, R. (September 2007). "Mutations in UPF3B, a member of the nonsense-mediated mRNA decay complex, cause syndromic and nonsyndromic mental retardation". Nature Genetics (Free full text). 39 (9): 1127–1133. doi:10.1038/ng2100. ISSN 1061-4036. PMC 2872770. PMID 17704778.
- Online Mendelian Inheritance in Man (OMIM): 305450
- Graham, J. M.; Superneau, D.; Rogers, R. C.; Corning, K.; Schwartz, C. E.; Dykens, E. M. (1999). "Clinical and behavioral characteristics in FG syndrome". American Journal of Medical Genetics. 85 (5): 470–475. doi:10.1002/(SICI)1096-8628(19990827)85:5<470::AID-AJMG7>3.0.CO;2-S. PMID 10405444.
- Graham, J. M. Jr.; Visootsak, J.; Dykens, E.; Huddleston, L.; Clark, R. D.; Jones, K. L.; Moeschler, J. B..; Opitz, J. M..; Morford, J.; Simensen, R.; Rogers, R. C.; Schwartz, C. E.; Friez, M. J.; Stevenson, R. E. (December 2008). "Behavior of 10 patients with FG Syndrome (Opitz-Kaveggia Syndrome) and the p.R961W Mutation in the MED12 Gene". American Journal of Medical Genetics Part A. 146A (23): 3011–3017. doi:10.1002/ajmg.a.32553. ISSN 1552-4825. PMC 3092600. PMID 18973276.
- Risheg, H.; Graham, J. M.; Clark, R. D.; Rogers, R. C.; Opitz, J. M.; Moeschler, J. B.; Peiffer, A. P.; May, M.; Joseph, S. M.; Jones, J. R.; Stevenson, R. E.; Schwartz, C. E.; Friez, M. J. (April 2007). "A recurrent mutation in MED12 leading to R961W causes Opitz-Kaveggia syndrome". Nature Genetics. 39 (4): 451–453. doi:10.1038/ng1992. ISSN 1061-4036. PMID 17334363. S2CID 26858160.
Further reading
- GeneReview/NIH/UW entry on MED12-Related Disorders
- Van Buggenhout, G. J. C. M.; Trommelen, J. C. M.; Brunner, H. G.; Hamel, B. C. J.; Fryns, J. P. (Jan 2001). "The clinical phenotype in institutionalised adult males with X-linked mental retardation (XLMR)". Annales de Génétique. 44 (1): 47–55. doi:10.1016/S0003-3995(01)01038-3. ISSN 0003-3995. PMID 11334618.