MFN2
Mitofusin-2 is a protein that in humans is encoded by the MFN2 gene.[5][6] Mitofusins are GTPases embedded in the outer membrane of the mitochondria. In mammals MFN1 and MFN2 are essential for mitochondrial fusion.[7] In addition to the mitofusins, OPA1 regulates inner mitochondrial membrane fusion, and DRP1 is responsible for mitochondrial fission.[8]




Mitofusin-2 (MFN2) is a mitochondrial membrane protein that plays a central role in regulating mitochondrial fusion and cell metabolism. More specifically, MFN2 is a dynamin-like GTPase embedded in the outer mitochondrial membrane (OMM) which in turn affects mitochondrial dynamics, distribution, quality control, and function.
In addition to the MFN2, OPA1 regulates inner mitochondrial membrane fusion, MFN1 is a mediator of mitochondrial fusion and DRP1 is responsible for mitochondrial fission.[8]
Structure
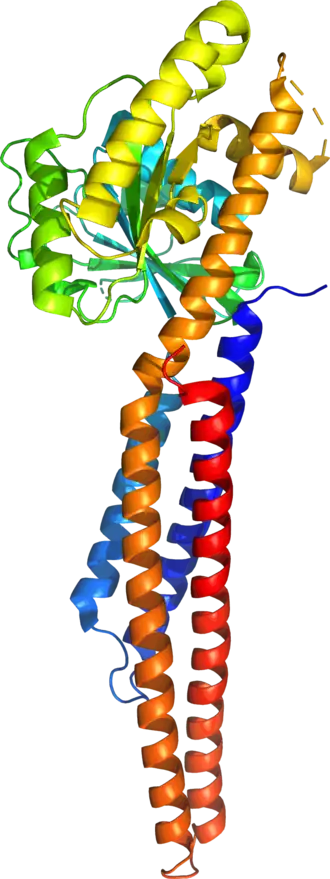
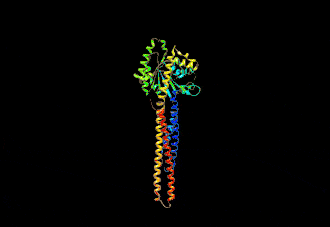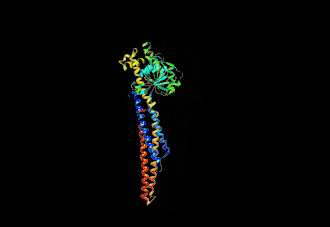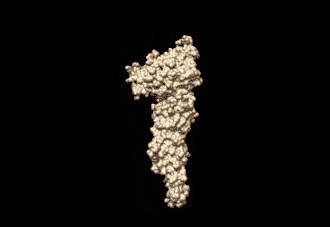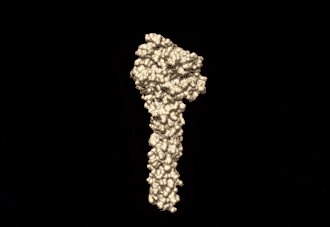
The human mitofusin-2 protein contains 757 amino acid residues. The MFN2 comprises a large cytosolic GTPase domain at the N-terminal, followed by a coiled-coil heptad-repeat (HR1) domain, a proline-rich (PR) region, two sequential transmembrane (TM) domains crossing the OMM and a second cytosolic heptad-repeat (HR2) domain at the C-terminal. MFN2 has been shown by electron microscopy (EM) to accumulate in contact regions between adjacent mitochondria, supporting their role in mitochondrial fusion.[10][11] Seminal studies revealed that both, MFN1 and MFN2 spanning from the OMM of two opposing mitochondria, physically interact in trans, by the formation of antiparallel dimers between their HR2 domains.[12]
Function
A pivotal in vivo study revealed that MFN2 is essential for embryonic development,[13] thus, the deletion of MFN2 in mice is lethal during midgestation. The inactivation of MFN2 alleles after placentation also revealed that MFN2 ablation severely impairs cerebellum development.[14] It has been also described that Mfn1 and Mfn2 are ubiquitously expressed yet they display different relative levels of expression between tissues, with MFN2 being the predominantly expressed mitofusin in the brain and MFN1 in the heart. This tissue-specific expression could be one of the reasons its ablation induces cerebellar-specific impairments.[15]
Mitochondrial fusion and fission
MFN2 is a mitochondrial membrane protein that participates in mitochondrial fusion and contributes to the maintenance and operation of the mitochondrial network.[16] Mitochondria function as a dynamic network constantly undergoing fusion and fission. The balance between fusion and fission is important in maintaining the integrity of the mitochondria and facilitates the mixing of the membranes and the exchange of DNA between mitochondria. MFN1 and MFN2 mediate outer membrane fusion, OPA1 is involved in inner membrane fusion, and DRP1 is responsible for mitochondrial fission.[17]
Mitochondrial fusion is unique because it involves two membranes: the OMM and the inner mitochondrial membrane (IMM), that must be rearranged in a coordinated manner in order to maintain organelle integrity.[15] Recent studies have shown that MFN2-deficient cells display an aberrant mitochondrial morphology, with a clear fragmentation of the network.[13]
Mitochondrial fusion is essential for embryonic development. Knockout mice for either MFN1 or MFN2 have fusion deficits and die midgestation. MFN2 knockout mice die at embryonic day 11.5 due to a defect in the giant cell layer of the placenta.[7] Mitochondrial fusion is also important for mitochondrial transport and localization in neuronal processes.[18] Conditional MFN2 knockout mice show degeneration in the Purkinje cells of the cerebellum, as well as improperly localized mitochondria in the dendrites.[19] MFN2 also associates with the MIRO-Milton complex which links the mitochondria to the kinesin motor.[18]
ER-mitochondria contacts
MFN2 has also been suggested to be a key regulator of ER-mitochondria contiguity, though its exact function in this inter-organelle still remains unknown. Small fractions of MFN2 have been observed to be located in ER membranes, particularly in the so called ER mitochondria-associated membranes (MAM).[19] Several processes known to take place at MAM, such as autophagosomes formation have been claimed to be modulated by the presence of MFN2.
Axonal transport of mitochondria
MFN2 has been proposed to be essential for the transport of mitochondria along axons, being involved in their attachment to microtubules through interaction with the two main motor proteins Miro and Milton.[20]
Other intracellular pathways, such as cell cycle progression, maintenance of mitochondrial bioenergetics, apoptosis, and autophagy, have been demonstrated to be modulated by MFN2.
Clinical significance
The importance of a regulated mitochondrial morphology in cell physiology makes immediately clear the potential impact of MFN2 in the onset/progression of different pathological conditions.[15]
Charcot–Marie–Tooth disease type 2A (CMT2A)
Charcot-Marie-Tooth disease type 2A (CMT2A) is caused by mutations in the MFN2 gene. MFN2 mutations are linked to neurological disorders characterized by a wide clinical phenotype that involves the central and peripheral nervous system.[21] [22] The impairment of the former is rarer while neuropathy forms are more frequent and severe, involving both legs and arms, with weakness, sensory loss, and optical atrophy.[21] All these complex phenotypes are clinically collected in the neurological disorder CMT2A, a subtype of a heterogeneous group of congenital neuromuscular diseases which affect motor and sensory neurons, called CMT disease.[23][24]
Among different cell types, neurons are particularly sensitive to MFN2 defects: to work properly, these cells need functional mitochondria located at specific sites to support adequate ATP production and Ca2+ buffering.[25] A defective mitochondrial fusion has been suggested to participate in the pathogenesis of CMT2A. Another important cell feature altered in the presence of MFN2 mutations is mitochondrial transport and indeed current models propose this defect as the major cause of CMT2A.
Mutations in OPA1 also cause optic atrophy, which suggests a common role of mitochondrial fusion in neuronal dysfunction.[19] The exact mechanism of how mutations in MFN2 selectively cause the degeneration of long peripheral axons is not known. There is evidence suggesting that it could be due to defects in the axonal transport of mitochondria.[19]
Alzheimer's disease
Increasing evidence suggests a possible link between MFN2 deregulation and Alzheimer's disease (AD). In particular, MFN2 protein and mRNA levels are decreased in the frontal cortex of patients with AD,[26] as well as in hippocampal neurons of post-mortem AD patients.[27] Notably, the cortex and hippocampus are the brain's areas in which a major neuronal impairment is observed in AD. Interestingly, the MFN2 gene is located on chromosome 1p36, which has been suggested to be an AD-associated locus.[28] However, it is currently unknown whether MFN2 alterations are causative for the pathology or just a consequence of AD. In particular, it is not clear if MFN2 is linked to AD through its effects on mitochondria or by affecting other pathways.
In summary, mitochondrial dysfunction is a prominent feature of AD neurons. It has been described that levels of DRP1 , OPA1, MFN1, and MFN2 are significantly reduced whereas levels of Fis1 are significantly increased in AD.[29]
Parkinson's disease
MFN2 is a key substrate of the PINK1/parkin couple, whose mutations are linked to the familial forms of Parkinson's disease (PD). MFN2 has been demonstrated to be essential for axonal projections of midbrain dopaminergic (DA) neurons that are affected in PD.[30] MFN2 alterations in the progression of PD, considering the capacity of PINK1 and parkin to trigger post-translational modifications in their substrates, have yet to be evaluated.
Obesity/diabetes/insulin resistance
The MFN2 protein may play a role in the pathophysiology of obesity.[31] In obesity and type II diabetes, MFN2 expression has been found to be reduced.[32][33] In turn, MFN2 down-regulation activates JNK pathway, favouring the formation of lipid intermediates that lead to insulin resistance. Recent studies have also shown that mitochondria arrest fusion by down-regulating MFN2 in obesity and diabetes, which leads to a fragmented mitochondrial network.[8] This fragmentation is obvious in the pancreatic beta-cells in the Islets of Langerhans and can inhibit mitochondrial quality control mechanisms such as mitophagy and autophagy - leading to a defect in insulin secretion and eventual beta-cell failure.[34] The expression of MFN2 in skeletal muscle is proportional to insulin sensitivity in this tissue,[35] and its expression is reduced in high-fat diet fed mice[36] and Zucker fatty rats.[35]
Cardiomyopathies
In heart, the embryonic combined MFN1/MFN2 deletion is lethal for mice embryo, while in adults it induces a progressive and lethal dilated cardiomyopathy.[37] A modest cardiac hypertrophy, associated to a tendency of MFN2-deprived mitochondria was observed caused by an increased resistance to Ca2+-mediated cell death stimuli.[38] While it is undisputed the importance of MFN2 in cardiomyocytes physiology, clarification of whether its pro-fusion activity or other functionalities of the protein are involved will require further investigations.
Cancer
Studying the mechanisms of mitochondrial function, more specifically MFN2 function, during tumorigenesis is critical for the next generation of cancer therapeutics. Recent studies have shown that dysregulation of the mitochondrial network can have an effect on MFN2 proteins, provoking mitochondrial hyperfusion and a multidrug resistant (MDR) phenotype in cancer cells.[39] MDR cancer cells have a much more aggressive behaviour and they are very invasive with a better ability to metastasize.[40] All these factors lead to a poor cancer prognosis and, therefore, novel therapeutic strategies for targeting and eradicating MDR TNBC cells are required. It has been hypothesized that mitochondrial hyperfusion is one of the main mechanisms that makes cells resistant to traditional chemotherapy treatments. Hence, inhibiting mitochondrial fusion would sensitize cancer cells to chemotherapy, making it a significantly more effective treatment. In order to inhibit mitochondrial hyperfusion, an anti-MFN2 peptide has to be used, in order to bind to the mitochondria membrane MFN2 proteins to prevent them from building the mitochondrial network.[41] The aim of the anti-MFN2 peptide is to make MFN2 not functional so it cannot participate in mitochondrial fusion and in the operation of the mitochondrial network. In this way, hyperfusion will not occur and chemotherapy drugs would be much more successful. However, further investigations are required in this field as there are still lots of unknowns.
References
- GRCh38: Ensembl release 89: ENSG00000116688 - Ensembl, May 2017
- GRCm38: Ensembl release 89: ENSMUSG00000029020 - Ensembl, May 2017
- "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- Karbowski M, Lee YJ, Gaume B, Jeong SY, Frank S, Nechushtan A, et al. (December 2002). "Spatial and temporal association of Bax with mitochondrial fission sites, Drp1, and Mfn2 during apoptosis". The Journal of Cell Biology. 159 (6): 931–8. doi:10.1083/jcb.200209124. PMC 2173996. PMID 12499352.
- Santel A, Fuller MT (March 2001). "Control of mitochondrial morphology by a human mitofusin". Journal of Cell Science. 114 (Pt 5): 867–74. doi:10.1242/jcs.114.5.867. PMID 11181170.
- Chan DC (June 2006). "Mitochondria: dynamic organelles in disease, aging, and development". Cell. 125 (7): 1241–52. doi:10.1016/j.cell.2006.06.010. PMID 16814712. S2CID 8551160.
- Liesa M, Shirihai OS (April 2013). "Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure". Cell Metabolism. 17 (4): 491–506. doi:10.1016/j.cmet.2013.03.002. PMC 5967396. PMID 23562075.
- PDB: 6JFL; Li YJ, Cao YL, Feng JX, Qi Y, Meng S, Yang JF, Zhong YT, Kang S, Chen X, Lan L, Luo L, Yu B, Chen S, Chan DC, Hu J, Gao S (October 2019). "Structural insights of human mitofusin-2 into mitochondrial fusion and CMT2A onset". Nature Communications. 10 (1): 4914. Bibcode:2019NatCo..10.4914L. doi:10.1038/s41467-019-12912-0. PMC 6820788. PMID 31664033.
- Rojo M, Legros F, Chateau D, Lombès A (April 2002). "Membrane topology and mitochondrial targeting of mitofusins, ubiquitous mammalian homologs of the transmembrane GTPase Fzo". Journal of Cell Science. 115 (Pt 8): 1663–74. doi:10.1242/jcs.115.8.1663. PMID 11950885.
- Santel A, Frank S, Gaume B, Herrler M, Youle RJ, Fuller MT (July 2003). "Mitofusin-1 protein is a generally expressed mediator of mitochondrial fusion in mammalian cells". Journal of Cell Science. 116 (Pt 13): 2763–74. doi:10.1242/jcs.00479. PMID 12759376. S2CID 6661619.
- Koshiba T, Detmer SA, Kaiser JT, Chen H, McCaffery JM, Chan DC (August 2004). "Structural basis of mitochondrial tethering by mitofusin complexes". Science. 305 (5685): 858–62. Bibcode:2004Sci...305..858K. doi:10.1126/science.1099793. PMID 15297672. S2CID 24595783.
- Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC (January 2003). "Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development". The Journal of Cell Biology. 160 (2): 189–200. doi:10.1083/jcb.200211046. PMC 2172648. PMID 12527753.
- Chen H, McCaffery JM, Chan DC (August 2007). "Mitochondrial fusion protects against neurodegeneration in the cerebellum". Cell. 130 (3): 548–62. doi:10.1016/j.cell.2007.06.026. PMID 17693261. S2CID 1138255.
- Filadi R, Pendin D, Pizzo P (February 2018). "Mitofusin 2: from functions to disease". Cell Death & Disease. 9 (3): 330. doi:10.1038/s41419-017-0023-6. PMC 5832425. PMID 29491355.
 Text was copied from this source, which is available under a Creative Commons Attribution 4.0 International License.
Text was copied from this source, which is available under a Creative Commons Attribution 4.0 International License.
- "Entrez Gene: MFN2 mitofusin 2".
- Chan DC (November 2006). "Dissecting mitochondrial fusion". Developmental Cell. 11 (5): 592–4. doi:10.1016/j.devcel.2006.10.009. PMID 17084350.
- Sheng ZH, Cai Q (January 2012). "Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration". Nature Reviews. Neuroscience. 13 (2): 77–93. doi:10.1038/nrn3156. PMC 4962561. PMID 22218207.
- Cartoni R, Martinou JC (August 2009). "Role of mitofusin 2 mutations in the physiopathology of Charcot-Marie-Tooth disease type 2A". Experimental Neurology. 218 (2): 268–73. doi:10.1016/j.expneurol.2009.05.003. PMID 19427854. S2CID 9341454.
- Misko A, Jiang S, Wegorzewska I, Milbrandt J, Baloh RH (March 2010). "Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex". The Journal of Neuroscience. 30 (12): 4232–40. doi:10.1523/jneurosci.6248-09.2010. PMC 2852190. PMID 20335458.
- Züchner S, De Jonghe P, Jordanova A, Claeys KG, Guergueltcheva V, Cherninkova S, et al. (February 2006). "Axonal neuropathy with optic atrophy is caused by mutations in mitofusin 2". Annals of Neurology. 59 (2): 276–81. doi:10.1002/ana.20797. PMID 16437557. S2CID 30679835.
- Vallat JM, Ouvrier RA, Pollard JD, Magdelaine C, Zhu D, Nicholson GA, et al. (November 2008). "Histopathological findings in hereditary motor and sensory neuropathy of axonal type with onset in early childhood associated with mitofusin 2 mutations". Journal of Neuropathology and Experimental Neurology. 67 (11): 1097–102. doi:10.1097/nen.0b013e31818b6cbc. PMID 18957892. S2CID 16302093.
- Cartoni R, Martinou JC (August 2009). "Role of mitofusin 2 mutations in the physiopathology of Charcot-Marie-Tooth disease type 2A". Experimental Neurology. 218 (2): 268–73. doi:10.1016/j.expneurol.2009.05.003. PMID 19427854. S2CID 9341454.
- Barisic N, Claeys KG, Sirotković-Skerlev M, Löfgren A, Nelis E, De Jonghe P, Timmerman V (May 2008). "Charcot-Marie-Tooth disease: a clinico-genetic confrontation". Annals of Human Genetics. 72 (Pt 3): 416–41. doi:10.1111/j.1469-1809.2007.00412.x. PMID 18215208. S2CID 33405406.
- Celsi F, Pizzo P, Brini M, Leo S, Fotino C, Pinton P, Rizzuto R (May 2009). "Mitochondria, calcium and cell death: a deadly triad in neurodegeneration". Biochimica et Biophysica Acta (BBA) - Bioenergetics. 1787 (5): 335–44. doi:10.1016/j.bbabio.2009.02.021. PMC 2696196. PMID 19268425.
- Manczak M, Calkins MJ, Reddy PH (July 2011). "Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer's disease: implications for neuronal damage". Human Molecular Genetics. 20 (13): 2495–509. doi:10.1093/hmg/ddr139. PMC 3109997. PMID 21459773.
- Chen Y, Han S, Huang X, Ni J, He X (January 2016). "The Protective Effect of Icariin on Mitochondrial Transport and Distribution in Primary Hippocampal Neurons from 3× Tg-AD Mice". International Journal of Molecular Sciences. 17 (2): 163. doi:10.3390/ijms17020163. PMC 4783897. PMID 26828481.
- Hiltunen M, Mannermaa A, Thompson D, Easton D, Pirskanen M, Helisalmi S, et al. (November 2001). "Genome-wide linkage disequilibrium mapping of late-onset Alzheimer's disease in Finland". Neurology. 57 (9): 1663–8. doi:10.1212/wnl.57.9.1663. PMID 11706108. S2CID 72375165.
- Wang X, Su B, Lee HG, Li X, Perry G, Smith MA, Zhu X (July 2009). "Impaired balance of mitochondrial fission and fusion in Alzheimer's disease". The Journal of Neuroscience. 29 (28): 9090–103. doi:10.1523/JNEUROSCI.1357-09.2009. PMC 2735241. PMID 19605646.
- Lee S, Sterky FH, Mourier A, Terzioglu M, Cullheim S, Olson L, Larsson NG (November 2012). "Mitofusin 2 is necessary for striatal axonal projections of midbrain dopamine neurons". Human Molecular Genetics. 21 (22): 4827–35. doi:10.1093/hmg/dds352. PMID 22914740.
- Zorzano A, Sebastián D, Segalés J, Palacín M (September 2009). "The molecular machinery of mitochondrial fusion and fission: An opportunity for drug discovery?". Current Opinion in Drug Discovery & Development. 12 (5): 597–606. PMID 19736619.
- Bach D, Naon D, Pich S, Soriano FX, Vega N, Rieusset J, et al. (September 2005). "Expression of Mfn2, the Charcot-Marie-Tooth neuropathy type 2A gene, in human skeletal muscle: effects of type 2 diabetes, obesity, weight loss, and the regulatory role of tumor necrosis factor alpha and interleukin-6". Diabetes. 54 (9): 2685–93. doi:10.2337/diabetes.54.9.2685. PMID 16123358.
- Kipanyula MJ, Contreras L, Zampese E, Lazzari C, Wong AK, Pizzo P, et al. (October 2012). "Ca2+ dysregulation in neurons from transgenic mice expressing mutant presenilin 2". Aging Cell. 11 (5): 885–93. doi:10.1111/j.1474-9726.2012.00858.x. PMID 22805202. S2CID 36750635.
- Trudeau KM, Colby AH, Zeng J, Las G, Feng JH, Grinstaff MW, Shirihai OS (July 2016). "Lysosome acidification by photoactivated nanoparticles restores autophagy under lipotoxicity". The Journal of Cell Biology. 214 (1): 25–34. doi:10.1083/jcb.201511042. PMC 4932370. PMID 27377248.
- Bach D, Pich S, Soriano FX, Vega N, Baumgartner B, Oriola J, et al. (May 2003). "Mitofusin-2 determines mitochondrial network architecture and mitochondrial metabolism. A novel regulatory mechanism altered in obesity". The Journal of Biological Chemistry. 278 (19): 17190–7. doi:10.1074/jbc.M212754200. PMID 12598526.
- Sorianello E, Soriano FX, Fernández-Pascual S, Sancho A, Naon D, Vila-Caballer M, et al. (April 2012). "The promoter activity of human Mfn2 depends on Sp1 in vascular smooth muscle cells". Cardiovascular Research. 94 (1): 38–47. doi:10.1093/cvr/cvs006. PMID 22253285.
- Chen Y, Liu Y, Dorn GW (December 2011). "Mitochondrial fusion is essential for organelle function and cardiac homeostasis". Circulation Research. 109 (12): 1327–31. doi:10.1161/circresaha.111.258723. PMC 3237902. PMID 22052916.
- Papanicolaou KN, Khairallah RJ, Ngoh GA, Chikando A, Luptak I, O'Shea KM, et al. (March 2011). "Mitofusin-2 maintains mitochondrial structure and contributes to stress-induced permeability transition in cardiac myocytes". Molecular and Cellular Biology. 31 (6): 1309–28. doi:10.1128/mcb.00911-10. PMC 3067905. PMID 21245373.
- Vyas S, Zaganjor E, Haigis MC (July 2016). "Mitochondria and Cancer". Cell. 166 (3): 555–566. doi:10.1016/j.cell.2016.07.002. PMC 5036969. PMID 27471965.
- Brown JM (2007). "Tumor hypoxia in cancer therapy". Oxygen Biology and Hypoxia. Methods in Enzymology. Vol. 435. Elsevier. pp. 297–321. doi:10.1016/s0076-6879(07)35015-5. ISBN 978-0-12-373970-4. PMID 17998060.
- Milane L, Trivedi M, Singh A, Talekar M, Amiji M (June 2015). "Mitochondrial biology, targets, and drug delivery". Journal of Controlled Release. 207: 40–58. doi:10.1016/j.jconrel.2015.03.036. PMID 25841699.
Further reading
- Pawlikowska P, Orzechowski A (2007). "[Role of transmembrane GTPases in mitochondrial morphology and activity]". Postepy Biochem. 53 (1): 53–9. PMID 17718388.
- Zorzano A, Bach D, Pich S, Palacín M (2004). "[Role of novel mitochondrial proteins in energy balance]". Revista de medicina de la Universidad de Navarra. 48 (2): 30–5. PMID 15382611.
External links
- GeneReviews/NCBI/NIH/UW entry on Charcot-Marie-Tooth Neuropathy Type 2
- Overview of all the structural information available in the PDB for UniProt: O95140 (Mitofusin-2) at the PDBe-KB.