MNase-seq
MNase-seq, short for micrococcal nuclease digestion with deep sequencing,[1][2][3][4] is a molecular biological technique that was first pioneered in 2006 to measure nucleosome occupancy in the C. elegans genome,[5] and was subsequently applied to the human genome in 2008.[2] Though, the term ‘MNase-seq’ had not been coined until a year later, in 2009.[3] Briefly, this technique relies on the use of the non-specific endo-exonuclease micrococcal nuclease, an enzyme derived from the bacteria Staphylococcus aureus, to bind and cleave protein-unbound regions of DNA on chromatin. DNA bound to histones or other chromatin-bound proteins (e.g. transcription factors) may remain undigested. The uncut DNA is then purified from the proteins and sequenced through one or more of the various Next-Generation sequencing methods.[6]
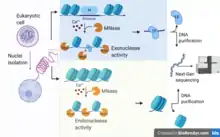
MNase-seq is one of four classes of methods used for assessing the status of the epigenome through analysis of chromatin accessibility. The other three techniques are DNase-seq, FAIRE-seq, and ATAC-seq.[4] While MNase-seq is primarily used to sequence regions of DNA bound by histones or other chromatin-bound proteins,[2] the other three are commonly used for: mapping Deoxyribonuclease I hypersensitive sites (DHSs),[7] sequencing the DNA unbound by chromatin proteins,[8] or sequencing regions of loosely packaged chromatin through transposition of markers,[9][10] respectively.[4]
History
Micrococcal nuclease (MNase) was first discovered in S. aureus in 1956,[11] protein crystallized in 1966,[12] and characterized in 1967.[13] MNase digestion of chromatin was key to early studies of chromatin structure; being used to determine that each nucleosomal unit of chromatin was composed of approximately 200bp of DNA.[14] This, alongside Olins’ and Olins’ “beads on a string” model,[15] confirmed Kornberg’s ideas regarding the basic chromatin structure.[16] Upon additional studies, it was found that MNase could not degrade histone-bound DNA shorter than ~140bp and that DNase I and II could degrade the bound DNA to as low as 10bp.[17][18] This ultimately elucidated that ~146bp of DNA wrap around the nucleosome core,[19] ~50bp linker DNA connect each nucleosome,[20] and that 10 continuous base-pairs of DNA tightly bind to the core of the nucleosome in intervals.[18]
In addition to being used to study chromatin structure, micrococcal nuclease digestion had been used in oligonucleotide sequencing experiments since its characterization in 1967.[21] MNase digestion was additionally used in several studies to analyze chromatin-free sequences, such as yeast (Saccharomyces cerevisiae) mitochondrial DNA[22] as well as bacteriophage DNA[23][24] through its preferential digestion of adenine and thymine-rich regions.[25] In the early 1980s, MNase digestion was used to determine the nucleosomal phasing and associated DNA for chromosomes from mature SV40,[26] fruit flies (Drosophila melanogaster),[27] yeast,[28] and monkeys,[29] among others. The first study to use this digestion to study the relevance of chromatin accessibility to gene expression in humans was in 1985. In this study, nuclease was used to find the association of certain oncogenic sequences with chromatin and nuclear proteins.[30] Studies utilizing MNase digestion to determine nucleosome positioning without sequencing or array information continued into the early 2000s.[31]
With the advent of whole genome sequencing in the late 1990s and early 2000s, it became possible to compare purified DNA sequences to the eukaryotic genomes of S. cerevisiae,[32] Caenorhabditis elegans,[33] D. melanogaster,[34] Arabidopsis thaliana,[35] Mus musculus,[36] and Homo sapiens.[37] MNase digestion was first applied to genome-wide nucleosome occupancy studies in S. cerevisiae[38] accompanied by analyses through microarrays to determine which DNA regions were enriched with MNase-resistant nucleosomes. MNase-based microarray analyses were often utilized at genome-wide scales for yeast[39][40] and in limited genomic regions in humans[41][42] to determine nucleosome positioning, which could be used as an inference for transcriptional inactivation.
In 2006, Next-Generation sequencing was first coupled with MNase digestion to explore nucleosome positioning and DNA sequence preferences in C. elegans,.[43] This was the first example of MNase-seq in any organism.
It was not until 2008, around the time Next-Generation sequencing was becoming more widely available, when MNase digestion was combined with high-throughput sequencing, namely Solexa/Illumina sequencing, to study nucleosomal positioning at a genome-wide scale in humans.[2] A year later, the terms “MNase-Seq” and “MNase-ChIP”, for micrococcal nuclease digestion with chromatin immunoprecipitation, were finally coined.[3] Since its initial application in 2006,[44] MNase-seq has been utilized to deep sequence DNA associated with nucleosome occupancy and epigenomics across eukaryotes.[6] As of February 2020, MNase-seq is still applied to assay accessibility in chromatin.[45]
Description
Chromatin is dynamic and the positioning of nucleosomes on DNA changes through the activity of various transcription factors and remodeling complexes, approximately reflecting transcriptional activity at these sites. DNA wrapped around nucleosomes are generally inaccessible to transcription factors.[46] Hence, MNase-seq can be used to indirectly determine which regions of DNA are transcriptionally inaccessible by directly determining which regions are bound to nucleosomes.[6]
In a typical MNase-seq experiment, eukaryotic cell nuclei are first isolated from a tissue of interest. Then, MNase-seq uses the endo-exonuclease micrococcal nuclease to bind and cleave protein-unbound regions of DNA of eukaryotic chromatin, first cleaving and resecting one strand, then cleaving the antiparallel strand as well.[3] The chromatin can be optionally crosslinked with formaldehyde.[47] MNase requires Ca2+ as a cofactor, typically with a final concentration of 1mM.[6][13] If a region of DNA is bound by the nucleosome core (i.e. histones) or other chromatin-bound proteins (e.g. transcription factors), then MNase is unable to bind and cleave the DNA. Nucleosomes or the DNA-protein complexes can be purified from the sample and the bound DNA can be subsequently purified via gel electrophoresis and extraction. The purified DNA is typically ~150bp, if purified from nucleosomes,[2] or shorter, if from another protein (e.g. transcription factors).[48] This makes short-read, high-throughput sequencing ideal for MNase-seq as reads for these technologies are highly accurate but can only cover a couple hundred continuous base-pairs in length.[49] Once sequenced, the reads can be aligned to a reference genome to determine which DNA regions are bound by nucleosomes or proteins of interest, with tools such as Bowtie.[4] The positioning of nucleosomes elucidated, through MNase-seq, can then be used to predict genomic expression[50] and regulation[51] at the time of digestion.
Extended Techniques
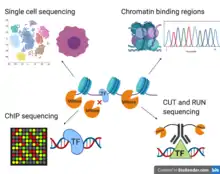
MNase-ChIP/CUT&RUN sequencing
Recently, MNase-seq has also been implemented in determining where transcription factors bind on the DNA.[52][53] Classical ChIP-seq displays issues with resolution quality, stringency in experimental protocol, and DNA fragmentation.[53] Classical ChIP-seq typically uses sonication to fragment chromatin, which biases heterochromatic regions due to the condensed and tight binding of chromatin regions to each other.[53] Unlike histones, transcription factors only transiently bind DNA. Other methods, such as sonication in ChIP-seq, requiring the use of increased temperatures and detergents, can lead to the loss of the factor. CUT&RUN sequencing is a novel form of an MNase-based immunoprecipitation. Briefly, it uses an MNase tagged with an antibody to specifically bind DNA-bound proteins that present the epitope recognized by that antibody. Digestion then specifically occurs at regions surrounding that transcription factor, allowing for this complex to diffuse out of the nucleus and be obtained without having to worry about significant background nor the complications of sonication. The use of this technique does not require high temperatures or high concentrations of detergent. Furthermore, MNase improves chromatin digestion due to its exonuclease and endonuclease activity. Cells are lysed in an SDS/Triton X-100 solution. Then, the MNase-antibody complex is added. And finally, the protein-DNA complex can be isolated, with the DNA being subsequently purified and sequenced. The resulting soluble extract contains a 25-fold enrichment in fragments under 50bp. This increased enrichment results in cost-effective high-resolution data.[53]
Single-cell MNase-seq
Single-cell micrococcal nuclease sequencing (scMNase-seq) is a novel technique that is used to analyze nucleosome positioning and to infer chromatin accessibility with the use of only a single-cell input.[54] First, cells are sorted into single aliquots using fluorescence-activated cell sorting (FACS).[54] The cells are then lysed and digested with micrococcal nuclease. The isolated DNA is subjected to PCR amplification and then the desired sequence is isolated and analyzed.[54] The use of MNase in single-cell assays results in increased detection of regions such as DNase I hypersensitive sites as well as transcription factor binding sites.[54]
Comparison to other Chromatin Accessibility Assays
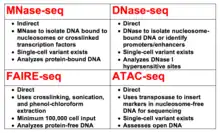
MNase-seq is one of four major methods (DNase-seq, MNase-seq, FAIRE-seq, and ATAC-seq) for more direct determination of chromatin accessibility and the subsequent consequences for gene expression.[55] All four techniques are contrasted with ChIP-seq, which relies on the inference that certain marks on histone tails are indicative of gene activation or repression,[56] not directly assessing nucleosome positioning, but instead being valuable for the assessment of histone modifier enzymatic function.[4]
DNase-seq
As with MNase-seq,[2] DNase-seq was developed by combining an existing DNA endonuclease[7] with Next-Generation sequencing technology to assay chromatin accessibility.[57] Both techniques have been used across several eukaryotes to ascertain information on nucleosome positioning in the respective organisms[4] and both rely on the same principle of digesting open DNA to isolate ~140bp bands of DNA from nucleosomes[2][58] or shorter bands if ascertaining transcription factor information.[48][58] Both techniques have recently been optimized for single-cell sequencing, which corrects for one of the major disadvantages of both techniques; that being the requirement for high cell input.[59][54]
At sufficient concentrations, DNase I is capable of digesting nucleosome-bound DNA to 10bp, whereas micrococcal nuclease cannot.[18] Additionally, DNase-seq is used to identify DHSs, which are regions of DNA that are hypersensitive to DNase treatment and are often indicative of regulatory regions (e.g. promoters or enhancers).[60] An equivalent effect is not found with MNase. As a result of this distinction, DNase-seq is primarily utilized to directly identify regulatory regions, whereas MNase-seq is used to identify transcription factor and nucleosomal occupancy to indirectly infer effects on gene expression.[4]
FAIRE-seq
FAIRE-seq differs more from MNase-seq than does DNase-seq.[4] FAIRE-seq was developed in 2007[8] and combined with Next-Generation sequencing three years later to study DHSs.[61] FAIRE-seq relies on the use of formaldehyde to crosslink target proteins with DNA and then subsequent sonication and phenol-chloroform extraction to separate non-crosslinked DNA and crosslinked DNA. The non-crosslinked DNA is sequenced and analyzed, allowing for direct observation of open chromatin.[62]
MNase-seq does not measure chromatin accessibility as directly as FAIRE-seq. However, unlike FAIRE-seq, it does not necessarily require crosslinking,[6] nor does it rely on sonication,[4] but it may require phenol and chloroform extraction.[6] Two major disadvantages of FAIRE-seq, relative to the other three classes, are the minimum required input of 100,000 cells and the reliance on crosslinking.[8] Crosslinking may bind other chromatin-bound proteins that transiently interact with DNA, hence limiting the amount of non-crosslinked DNA that can be recovered and assayed from the aqueous phase.[55] Thus, the overall resolution obtained from FAIRE-seq can be relatively lower than that of DNase-seq or MNase-seq[55] and with the 100,000 cell requirement,[8] the single-cell equivalents of DNase-seq[59] or MNase-seq[54] make them far more appealing alternatives.[4]
ATAC-seq
ATAC-seq is the most recently developed class of chromatin accessibility assays.[9] ATAC-seq uses a hyperactive transposase to insert transposable markers with specific adapters, capable of binding primers for sequencing, into open regions of chromatin. PCR can then be used to amplify sequences adjacent to the inserted transposons, allowing for determination of open chromatin sequences without causing a shift in chromatin structure.[9][10] ATAC-seq has been proven effective in humans, amongst other eukaryotes, including in frozen samples.[63] As with DNase-seq[59] and MNase-seq,[54] a successful single-cell version of ATAC-seq has also been developed.[64]
ATAC-seq has several advantages over MNase-seq in assessing chromatin accessibility. ATAC-seq does not rely on the variable digestion of the micrococcal nuclease, nor crosslinking or phenol-chloroform extraction.[6][10] It generally maintains chromatin structure, so results from ATAC-seq can be used to directly assess chromatin accessibility, rather than indirectly via MNase-seq. ATAC-seq can also be completed within a few hours,[10] whereas the other three techniques typically require overnight incubation periods.[6][7][8] The two major disadvantages to ATAC-seq, in comparison to MNase-seq, are the requirement for higher sequencing coverage and the prevalence of mitochondrial contamination due to non-specific insertion of DNA into both mitochondrial DNA and nuclear DNA.[9][10] Despite these minor disadvantages, use of ATAC-seq over the alternatives is becoming more prevalent.[4]
References
- Johnson SM, Tan FJ, McCullough HL, Riordan DP, Fire AZ (December 2006). "Flexibility and constraint in the nucleosome core landscape of Caenorhabditis elegans chromatin". Genome Research. 16 (12): 1505–16. doi:10.1101/gr.5560806. PMC 1665634. PMID 17038564.
- Schones DE, Cui K, Cuddapah S, Roh TY, Barski A, Wang Z, et al. (March 2008). "Dynamic regulation of nucleosome positioning in the human genome". Cell. 132 (5): 887–98. doi:10.1016/j.cell.2008.02.022. PMID 18329373. S2CID 13320420.
- Kuan PF, Huebert D, Gasch A, Keles S (January 2009). "A non-homogeneous hidden-state model on first order differences for automatic detection of nucleosome positions". Statistical Applications in Genetics and Molecular Biology. 8 (1): Article29. doi:10.2202/1544-6115.1454. PMC 2861327. PMID 19572828.
- Klein DC, Hainer SJ (November 2019). "Genomic methods in profiling DNA accessibility and factor localization". Chromosome Research. 28 (1): 69–85. doi:10.1007/s10577-019-09619-9. PMC 7125251. PMID 31776829.
- Johnson SM, Tan FJ, McCullough HL, Riordan DP, Fire AZ (December 2006). "Flexibility and constraint in the nucleosome core landscape of Caenorhabditis elegans chromatin". Genome Research. 16 (12): 1505–16. doi:10.1101/gr.5560806. PMC 1665634. PMID 17038564.
- Cui K, Zhao K (January 2012). "Genome-wide approaches to determining nucleosome occupancy in metazoans using MNase-Seq". Chromatin Remodeling. Methods in Molecular Biology. Vol. 833. pp. 413–9. doi:10.1007/978-1-61779-477-3_24. ISBN 978-1-61779-476-6. PMC 3541821. PMID 22183607.
- Giresi PG, Kim J, McDaniell RM, Iyer VR, Lieb JD (June 2007). "FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin". Genome Research. 17 (6): 877–85. doi:10.1101/gr.5533506. PMC 3959825. PMID 17179217.
- Giresi PG, Kim J, McDaniell RM, Iyer VR, Lieb JD (June 2007). "FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin". Genome Research. 17 (6): 877–85. doi:10.1101/gr.5533506. PMC 1891346. PMID 17179217.
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ (December 2013). "Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position". Nature Methods. 10 (12): 1213–8. doi:10.1038/nmeth.2688. PMC 3959825. PMID 24097267.
- Buenrostro JD, Wu B, Chang HY, Greenleaf WJ (January 2015). "ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide". Current Protocols in Molecular Biology. 109: 21.29.1–21.29.9. doi:10.1002/0471142727.mb2129s109. PMC 4374986. PMID 25559105.
- Cunningham L, Catlin BW, De Garilhe MP (September 1956). "A deoxyribonuclease of Micrococcus pyogenes". Journal of the American Chemical Society. 78 (18): 4642–4645. doi:10.1021/ja01599a031.
- Cotton FA, Hazen EE, Richardson DC (October 1966). "Crystalline extracellular nuclease of Staphylococcus aureus". The Journal of Biological Chemistry. 241 (19): 4389–90. doi:10.1016/S0021-9258(18)99732-2. PMID 5922963.
- Heins JN, Suriano JR, Taniuchi H, Anfinsen CB (March 1967). "Characterization of a nuclease produced by Staphylococcus aureus". The Journal of Biological Chemistry. 242 (5): 1016–20. doi:10.1016/S0021-9258(18)96225-3. PMID 6020427.
- Noll M (September 1974). "Subunit structure of chromatin". Nature. 251 (5472): 249–51. Bibcode:1974Natur.251..249N. doi:10.1038/251249a0. PMID 4422492. S2CID 637383.
- Olins AL, Olins DE (January 1974). "Spheroid chromatin units (v bodies)". Science. 183 (4122): 330–2. Bibcode:1974Sci...183..330O. doi:10.1126/science.183.4122.330. PMID 4128918. S2CID 83480762.
- Kornberg RD (May 1974). "Chromatin structure: a repeating unit of histones and DNA". Science. 184 (4139): 868–71. Bibcode:1974Sci...184..868K. doi:10.1126/science.184.4139.868. PMID 4825889.
- Keichline LD, Villee CA, Wassarman PM (February 1976). "Structure of eukaryotic chromatin. Evaluation of periodicity using endogenous and exogenous nucleases". Biochimica et Biophysica Acta (BBA) - Nucleic Acids and Protein Synthesis. 425 (1): 84–94. doi:10.1016/0005-2787(76)90218-5. PMID 1247619.
- Duerksen JD, Connor KW (April 1978). "Periodicity and fragment size of DNA from mouse TLT hepatoma chromatin and chromatin fractions using endogenous and exogenous nucleases". Molecular and Cellular Biochemistry. 19 (2): 93–112. doi:10.1007/bf00232599. PMID 206820. S2CID 9230112.
- Kornberg RD, Lorch Y (August 1999). "Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome". Cell. 98 (3): 285–94. doi:10.1016/s0092-8674(00)81958-3. PMID 10458604. S2CID 14039910.
- Whitlock JP, Simpson RT (July 1976). "Removal of histone H1 exposes a fifty base pair DNA segment between nucleosomes". Biochemistry. 15 (15): 3307–14. doi:10.1021/bi00660a022. PMID 952859.
- Feldmann H (July 1967). "[Sequence analysis of oliogonucleotides by means of micrococcal nuclease]". European Journal of Biochemistry. 2 (1): 102–5. doi:10.1111/j.1432-1033.1967.tb00113.x. PMID 6079759.
- Prunell A, Bernardi G (July 1974). "The mitochondrial genome of wild-type yeast cells. IV. Genes and spacers". Journal of Molecular Biology. 86 (4): 825–41. doi:10.1016/0022-2836(74)90356-8. PMID 4610147.
- Barrell BG, Weith HL, Donelson JE, Robertson HD (March 1975). "Sequence analysis of the ribosome-protected bacteriophase phiX174 DNA fragment containing the gene G initiation site". Journal of Molecular Biology. 92 (3): 377–93. doi:10.1016/0022-2836(75)90287-9. PMID 1095758.
- Bambara R, Wu R (June 1975). "DNA sequence analysis. Terminal sequences of bacteriophage phi80". The Journal of Biological Chemistry. 250 (12): 4607–18. doi:10.1016/S0021-9258(19)41345-8. PMID 166999.
- Wingert L, Von Hippel PH (March 1968). "The conformation dependent hydrolysis of DNA by micrococcal nuclease". Biochimica et Biophysica Acta (BBA) - Nucleic Acids and Protein Synthesis. 157 (1): 114–26. doi:10.1016/0005-2787(68)90270-0. PMID 4296058.
- Hiwasa T, Segawa M, Yamaguchi N, Oda K (May 1981). "Phasing of nucleosomes in SV40 chromatin reconstituted in vitro". Journal of Biochemistry. 89 (5): 1375–89. doi:10.1093/oxfordjournals.jbchem.a133329. PMID 6168635.
- Samal B, Worcel A, Louis C, Schedl P (February 1981). "Chromatin structure of the histone genes of D. melanogaster". Cell. 23 (2): 401–9. doi:10.1016/0092-8674(81)90135-5. PMID 6258802. S2CID 42138156.
- Lohr DE (October 1981). "Detailed analysis of the nucleosomal organization of transcribed DNA in yeast chromatin". Biochemistry. 20 (21): 5966–72. doi:10.1021/bi00524a007. PMID 6272832.
- Musich PR, Brown FL, Maio JJ (January 1982). "Nucleosome phasing and micrococcal nuclease cleavage of African green monkey component alpha DNA". Proceedings of the National Academy of Sciences of the United States of America. 79 (1): 118–22. Bibcode:1982PNAS...79..118M. doi:10.1073/pnas.79.1.118. PMC 345673. PMID 6275381.
- Kasid UN, Hough C, Thraves P, Dritschilo A, Smulson M (April 1985). "The association of human c-Ha-ras sequences with chromatin and nuclear proteins". Biochemical and Biophysical Research Communications. 128 (1): 226–32. doi:10.1016/0006-291x(85)91668-7. PMID 3885946.
- Goriely S, Demonté D, Nizet S, De Wit D, Willems F, Goldman M, Van Lint C (June 2003). "Human IL-12(p35) gene activation involves selective remodeling of a single nucleosome within a region of the promoter containing critical Sp1-binding sites". Blood. 101 (12): 4894–902. doi:10.1182/blood-2002-09-2851. PMID 12576336.
- Goffeau A, Barrell BG, Bussey H, Davis RW, Dujon B, Feldmann H, et al. (October 1996). "Life with 6000 genes". Science. 274 (5287): 546, 563–7. Bibcode:1996Sci...274..546G. doi:10.1126/science.274.5287.546. PMID 8849441. S2CID 16763139.
- The C. Elegans Sequencing Consortium (December 1998). "Genome sequence of the nematode C. elegans: a platform for investigating biology". Science. 282 (5396): 2012–8. Bibcode:1998Sci...282.2012.. doi:10.1126/science.282.5396.2012. PMID 9851916.
- Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG, et al. (March 2000). "The genome sequence of Drosophila melanogaster". Science. 287 (5461): 2185–95. Bibcode:2000Sci...287.2185.. doi:10.1126/science.287.5461.2185. PMID 10731132.
- The Arabidopsis Genome Initiative; et al. (The Arabidopsis Genome Initiative) (December 2000). "Analysis of the genome sequence of the flowering plant Arabidopsis thaliana". Nature. 408 (6814): 796–815. Bibcode:2000Natur.408..796T. doi:10.1038/35048692. PMID 11130711.
- Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, et al. (Mouse Genome Sequencing Consortium) (December 2002). "Initial sequencing and comparative analysis of the mouse genome". Nature. 420 (6915): 520–62. Bibcode:2002Natur.420..520W. doi:10.1038/nature01262. PMID 12466850.
- International Human Genome Sequencing Consortium (October 2004). "Finishing the euchromatic sequence of the human genome". Nature. 431 (7011): 931–45. Bibcode:2004Natur.431..931H. doi:10.1038/nature03001. PMID 15496913.
- Bernstein BE, Liu CL, Humphrey EL, Perlstein EO, Schreiber SL (August 2004). "Global nucleosome occupancy in yeast". Genome Biology. 5 (9): R62. doi:10.1186/gb-2004-5-9-r62. PMC 522869. PMID 15345046.
- Yuan GC, Liu YJ, Dion MF, Slack MD, Wu LF, Altschuler SJ, Rando OJ (July 2005). "Genome-scale identification of nucleosome positions in S. cerevisiae" (PDF). Science. 309 (5734): 626–630. Bibcode:2005Sci...309..626Y. doi:10.1126/science.1112178. PMID 15961632. S2CID 43625066.
- Lee W, Tillo D, Bray N, Morse RH, Davis RW, Hughes TR, Nislow C (October 2007). "A high-resolution atlas of nucleosome occupancy in yeast". Nature Genetics. 39 (10): 1235–44. doi:10.1038/ng2117. PMID 17873876. S2CID 12816925.
- Ozsolak F, Song JS, Liu XS, Fisher DE (February 2007). "High-throughput mapping of the chromatin structure of human promoters". Nature Biotechnology. 25 (2): 244–8. doi:10.1038/nbt1279. PMID 17220878. S2CID 365969.
- Dennis JH, Fan HY, Reynolds SM, Yuan G, Meldrim JC, Richter DJ, et al. (June 2007). "Independent and complementary methods for large-scale structural analysis of mammalian chromatin". Genome Research. 17 (6): 928–39. doi:10.1101/gr.5636607. PMC 1891351. PMID 17568008.
- Johnson SM, Tan FJ, McCullough HL, Riordan DP, Fire AZ (December 2006). "Flexibility and constraint in the nucleosome core landscape of Caenorhabditis elegans chromatin". Genome Research. 16 (12): 1505–16. doi:10.1101/gr.5560806. PMC 1665634. PMID 17038564.
- Johnson SM, Tan FJ, McCullough HL, Riordan DP, Fire AZ (December 2006). "Flexibility and constraint in the nucleosome core landscape of Caenorhabditis elegans chromatin". Genome Research. 16 (12): 1505–16. doi:10.1101/gr.5560806. PMC 1665634. PMID 17038564.
- Zhao H, Zhang W, Zhang T, Lin Y, Hu Y, Fang C, Jiang J (February 2020). "Genome-wide MNase hypersensitivity assay unveils distinct classes of open chromatin associated with H3K27me3 and DNA methylation in Arabidopsis thaliana". Genome Biology. 21 (1): 24. doi:10.1186/s13059-020-1927-5. PMC 6996174. PMID 32014062.
- Hargreaves DC, Crabtree GR (March 2011). "ATP-dependent chromatin remodeling: genetics, genomics and mechanisms". Cell Research. 21 (3): 396–420. doi:10.1038/cr.2011.32. PMC 3110148. PMID 21358755.
- Mieczkowski J, Cook A, Bowman SK, Mueller B, Alver BH, Kundu S, et al. (May 2016). "MNase titration reveals differences between nucleosome occupancy and chromatin accessibility". Nature Communications. 7: 11485. Bibcode:2016NatCo...711485M. doi:10.1038/ncomms11485. PMC 4859066. PMID 27151365.
- Hainer SJ, Fazzio TG (October 2015). "Regulation of Nucleosome Architecture and Factor Binding Revealed by Nuclease Footprinting of the ESC Genome". Cell Reports. 13 (1): 61–69. doi:10.1016/j.celrep.2015.08.071. PMC 4598306. PMID 26411677.
- Liu L, Li Y, Li S, Hu N, He Y, Pong R, et al. (January 2012). "Comparison of next-generation sequencing systems". Journal of Biomedicine & Biotechnology. 2012: 251364. doi:10.1155/2012/251364. PMC 3398667. PMID 22829749.
- Henikoff S (January 2008). "Nucleosome destabilization in the epigenetic regulation of gene expression". Nature Reviews. Genetics. 9 (1): 15–26. doi:10.1038/nrg2206. PMID 18059368. S2CID 24413271.
- Ercan S, Carrozza MJ, Workman JL (September 2004). "Global nucleosome distribution and the regulation of transcription in yeast". Genome Biology. 5 (10): 243. doi:10.1186/gb-2004-5-10-243. PMC 545588. PMID 15461807.
- Gutin J, Sadeh R, Bodenheimer N, Joseph-Strauss D, Klein-Brill A, Alajem A, et al. (March 2018). "Fine-Resolution Mapping of TF Binding and Chromatin Interactions". Cell Reports. 22 (10): 2797–2807. doi:10.1016/j.celrep.2018.02.052. PMC 5863041. PMID 29514105.
- Skene PJ, Henikoff S (June 2015). "A simple method for generating high-resolution maps of genome-wide protein binding". eLife. 4: e09225. doi:10.7554/eLife.09225. PMC 4480131. PMID 26079792.
- Lai B, Gao W, Cui K, Xie W, Tang Q, Jin W, et al. (October 2018). "Principles of nucleosome organization revealed by single-cell micrococcal nuclease sequencing". Nature. 562 (7726): 281–285. Bibcode:2018Natur.562..281L. doi:10.1038/s41586-018-0567-3. PMC 8353605. PMID 30258225. S2CID 52841785.
- Tsompana M, Buck MJ (November 2014). "Chromatin accessibility: a window into the genome". Epigenetics & Chromatin. 7 (1): 33. doi:10.1186/1756-8935-7-33. PMC 4253006. PMID 25473421.
- Park PJ (October 2009). "ChIP-seq: advantages and challenges of a maturing technology". Nature Reviews. Genetics. 10 (10): 669–80. doi:10.1038/nrg2641. PMC 3191340. PMID 19736561.
- Boyle AP, Davis S, Shulha HP, Meltzer P, Margulies EH, Weng Z, et al. (January 2008). "High-resolution mapping and characterization of open chromatin across the genome". Cell. 132 (2): 311–22. doi:10.1016/j.cell.2007.12.014. PMC 2669738. PMID 18243105.
- He HH, Meyer CA, Hu SS, Chen MW, Zang C, Liu Y, et al. (January 2014). "Refined DNase-seq protocol and data analysis reveals intrinsic bias in transcription factor footprint identification". Nature Methods. 11 (1): 73–78. doi:10.1038/nmeth.2762. PMC 4018771. PMID 24317252.
- Cooper J, Ding Y, Song J, Zhao K (November 2017). "Genome-wide mapping of DNase I hypersensitive sites in rare cell populations using single-cell DNase sequencing". Nature Protocols. 12 (11): 2342–2354. doi:10.1038/nprot.2017.099. PMID 29022941. S2CID 7993995.
- Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, et al. (September 2012). "The accessible chromatin landscape of the human genome". Nature. 489 (7414): 75–82. Bibcode:2012Natur.489...75T. doi:10.1038/nature11232. PMC 2828505. PMID 22955617.
- Gaulton KJ, Nammo T, Pasquali L, Simon JM, Giresi PG, Fogarty MP, et al. (March 2010). "A map of open chromatin in human pancreatic islets". Nature Genetics. 42 (3): 255–9. doi:10.1038/ng.530. PMC 2828505. PMID 20118932.
- Simon JM, Giresi PG, Davis IJ, Lieb JD (January 2012). "Using formaldehyde-assisted isolation of regulatory elements (FAIRE) to isolate active regulatory DNA". Nature Protocols. 7 (2): 256–67. doi:10.1038/nprot.2011.444. PMC 3784247. PMID 22262007.
- Corces MR, Buenrostro JD, Wu B, Greenside PG, Chan SM, Koenig JL, et al. (October 2016). "Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution". Nature Genetics. 48 (10): 1193–203. doi:10.1038/ng.3646. PMC 5042844. PMID 27526324.
- Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, et al. (July 2015). "Single-cell chromatin accessibility reveals principles of regulatory variation". Nature. 523 (7561): 486–90. Bibcode:2015Natur.523..486B. doi:10.1038/nature14590. PMC 4685948. PMID 26083756.