Meta-selective C–H functionalization
Meta-selective C–H functionalization refers to the regioselective reaction of a substituted aromatic ring on the C–H bond meta to the substituent.
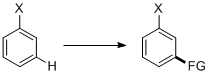
Substituted aromatic ring is an important type of substructure in pharmaceuticals and industrial compounds. Thus, synthetic methods towards substituted aromatic rings are always of great interest to chemists.
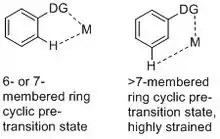
Traditionally, regioselectivity on the aromatic ring is achieved by the electronic effect of substituents. Taking the well-known Friedel–Craft electrophilic aromatic substitution as example, electron donating groups direct the electrophile to ortho-/para-position while electron withdrawing groups direct the electrophile to meta-position. However, with complicated systems, electronic difference between different C–H bonds can be subtle and electronic directing effect alone could become less synthetically useful. The fast development of C–H activation in the past few decades provides synthetic chemists with the powerful tools to synthesize functionalized aromatic compounds with high selectivity. The widely used approach to achieve ortho-selectivity involves metal-chelating directing groups, which forms a relatively stable 6- or 7-membered cyclic pre-transition state to bring the metal catalyst to the proximity of the ortho-hydrogen.[1][2][3][4][5] However, applying the same strategy to meta- or para- C-H functionalization does not work because the corresponding cyclophane-like cyclic pre-transition state is highly strained.[6] Thus, while ortho-selectivity has been achieved by numerous catalytic systems, meta- and para-selectivity remains a challenge.
In recent years, new strategies that override the electronic and steric bias have been developed to address meta-C–H functionalization. However, before these discoveries, synthesis of meta-substituted aromatic compounds could be either limited or cumbersome. For example, before the development of the C–H activation involving one-pot synthetic route to meta-substituted phenol derivatives by Maleczka and co-workers, the traditional synthesis requires 10 steps from TNT.[7][8][9] Some early attempts utilize steric and electronic effects to achieve meta-selectivity.[10][11][12] However, they are either limited to certain structure of substrates or are not highly selective. In recent years, several highly selective meta-C-H functionalization strategies have been reported which can override the intrinsic electronic and steric properties of the substrates and can apply to a wide range of substrate derivatives. The development of the modern meta-C-H functionalization strategies “open doors for numerous possibilities” for synthesis and catalyst development.[13]


Recently-developed meta-selective C–H activation strategies
Copper catalyzed meta-selective C–H arylation

In 2009, Gaunt's group reported a copper catalyzed meta-selective C–H arylation reaction on anilide derivatives.[14] Despite the intrinsic ortho-/para- selectivity of the amido group, the arylation occurs exclusively on the meta position on a variety of anilide substrates. Remarkably, the regioselectivity is totally different from the earlier reported Pd catalyzed C-H functionalizations, where the amido group serve as a powerful ortho-directing group.[7][15][16] The method is robust under mild reaction conditions. It is compatible with a spectrum of substituted anilide as well as different bisaryliodonium salts. However, the meta-selectivity is lost when highly ortho/para-directing methoxy group substitutes one of the meta-hydrogen of the anilide, which marks the limitation of this method.

Despite the limitation, the paper was of high impact. It has been highlighted in a number of journals and news and was voted as one of the top 12 papers of 2009 by Chemical and Engineering News.[17] In a more recent report from the same group, α-arylcarbonyl compounds were found to be good substrates for the copper catalyzed meta-selective C-H arylation.[18] The power of the meta-selectivity overrides the electronic effect of different substituents, including the strong ortho/para-directing m-methoxy group.

Although the copper catalyzed meta-selective C–H arylation is quite successful, the mechanism behind the meta-selectivity is not completely understood. There are generally two proposed mechanisms both involving a Cu(I)/Cu(III) catalyst cycle. In Gaunt's original paper, he proposed a mechanism involving an anti-oxy-cupration step as the key to the meta-selectivity.[14] First, the Cu(II) salt generates the active Cu(I) species through either disproportionation or reduction by nucleophile.[19] The active Cu(I) species undergoes oxidative addition with diphenyliodonium salt to generate a highly electrophilic Cu(III) species.[14][20] While the Cu(III) species activates the aromatic ring,[14] the amide oxygen attacks the ortho position, breaking the aromaticity and allowing cupration at the meta position.[14][21] The intermediate then rearomatizes with base and undergoes reductive elimination to afford the meta-arylated product and regenerate the active Cu(I) catalyst.[14]

Alternatively, Li and Wu, based on DFT calculations, proposed a mechanism involving a "Heck-like four-membered-ring transition state".[19] The amide oxygen first coordinates to the Cu(III) species generated from oxidative addition of Cu(I) triflate and diphenyliodonium triflate. Then, the phenyl group bonded to copper interacts with the aromatic ring at the meta-position, forming a four-membered-ring transition state. According to their calculations, the aromaticity is not completely lost during the transformation. In the last step, the Cu(III)-C bond breaks to regenerate the Cu(I) catalyst while the triflate ion abstract the meta-hydrogen to recover the aromaticity and gives the product.[19]

Meta-selective C-H functionalization assisted by a remote "end-on" template

In 2012, Yu and co-workers reported a pioneering meta-selective C-H olefination using nitrile-containing templates to deliver the palladium to the meta-position via a macrocyclic cyclophane-like pre-transition state.[6] The nitrile group is tethered to the aromatic ring by a removable linker. It coordinates weakly to palladium in an "end-on" fashion, which refers to the linear structure of C–CN–Pd bonds.[22]
The linear coordination is proposed to help overcome the high strain in the cyclophane-like pre-transition state that brings palladium to the vicinity of the meta-hydrogen.[6] The template is designed as such that the flat arene linker keeps the substrate aromatic ring and the nitrile group coplanar. Bulky substituents on the arene linker "lock" the nitrile tether in the desired position. The delicate design results in high regioselectivity towards the meta-C-H bond. The templates can be removed easily to give toluene derivatives or hydrocinnamic acid derivatives in high yield.
In their subsequent works, Yu and co-workers report the application of the same strategy in meta-selective C-H cross-coupling,[23] meta-C-H acetoxylation and meta-C–H olefination[24] in a broad substrate scope. It is demonstrated that the "end-on" template not only work with the Pd(0)/Pd(II) catalytic cycle but is also compatible with the Pd(II)/Pd(IV) cycle.[24] In all three works, addition of mono-N-protected amino acid (MPAA) such as N-acetyl glycine improves the reaction yields and enhances the regioselectivity.[6][23][24][25]
Mechanism
The mechanistic study of the palladium-catalyzed meta-selective C–H bond activation with a nitrile-containing template was done by Yu, Wu, Houk and their co-workers.[25] The DFT calculations suggest that the regioselectivity is achieved in the C–H activation step, which is the rate-determining step. It proceeds via a concerted metalation-deprotonation (CMD) pathway, which means that palladation and deprotonation of the C–H bond happen at the same time. Surprisingly, calculations reveal that the Pd–Ag heterodimeric transition state leads to meta-selectivity while the Pd monomeric transition state leads to ortho-selectivity.
The role of mono-N-protected amino acid is proposed as a dianionic ligand which participates in the CMD step assisting the deprotonation of the C–H bond in the rate- and regio-determining step.[25]
Meta-alkylation achieved by remote ortho-ruthenation and electrophilic-type substitution
Originally discovered by Frost and co-workers, the meta selective sulfonation of 2-phenylpyridine using a sulfonyl chloride coupling partner,[26] utilising a ruthenium(II) catalyst. This reaction has been proposed to proceed via a similar method to that of the meta alkylation reported by Frost and Ackermann which involves a meta-selective C-H bond alkylation reaction with secondary and tertiary alkyl halides catalyzed by ruthenium(II) carboxylate catalysts.[27][28] The directing group first coordinate to the ruthenium catalyst. A reversible metalation takes place to generate the cycloruthenated complex as the key intermediates. The cycloruthenation activates the aromatic ring to undergo SEAr type alkylation at the position para to the C–Ru bond.

Potential applications
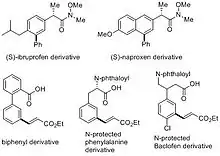
In Gaunt's and Yu's works, some derivatives of drug molecules and biologically active compounds were successfully functionalized in their meta-position. For instance, meta-arylated derivatives of anti-inflammatory drugs (S)-ibruprofen and (S)-naproxen were synthesized with copper catalyzed C–H arylation.[18] Meta-olefinated biologically important biphenyl, amino acid and Baclofen derivatives have been accessed by remote C–H activation assisted by the "end-on" template.[6] These demonstrate the potential applications of meta-selective C–H functionalization in medicinal chemistry.
See also
References
- Hartung, C. G.; Snieckus, V. in Modern Arene Chemistry (ed. Astruc, D.) 330–367 (Wiley-VCH, 2004).
- Colby, D. A., Bergman, R. G.&Ellman, J. A. Chem. Rev. 2010, 110, 624–655.
- Lyons, T. W. & Sanford, M. S. Chem. Rev. 2010, 110, 1147–1169.
- Jun, C. H.; Moon, C. W.; Lee, D-Y. Chem. Eur. J. 2002, 8 2422 – 2428.
- Engle, K. M.; Mei, T-S.; Wasa, M.; Yu, J-Q. Acc. Chem. Res., 2012, 45, 788–802.
- Leow, D.; Li, G.; Mei, T-S.; Yu, J-Q. Nature 2012, 486, 518.
- Boele, M. D. K.; van Strijdonck, G. P. F.; de Vries, A. H. M.; Kamer, P. C. J.; de Vries, J. G.; van Leeuwen, P. W. N. M. J. Am. Chem. Soc. 2002, 124, 1586.
- Hodgson, H. H.; Wignall, J. S. J. Chem. Soc. 1926, 129, 2077.
- Prakash Kumar Shee, Meta-Selective C-H Bond Activation. Department of Chemistry, Michigan State University, November 27, 2013, https://www2.chemistry.msu.edu/Courses/CEM958/FS13_SS14/Prakash_Kumar_Shee.pdf retrieved November 27, 2014
- Cho, J.-Y.; Tse, M. K.; Holmes, D.; Maleczka, R. E., Jr.; Smith, M. R., III Science 2002, 295, 305.
- Murphy, J. M.; Liao, X.; Hartwig, J. F. J. Am. Chem. Soc. 2007, 129, 15434.
- Zhang, Y-H.; Shi, B-F.; Yu, J-Q. J. Am. Chem. Soc. 2009, 131, 5072 – 5074.
- Zhou, Y.; Zhao, J.; Liu, L. Angew. Chem. Int. Ed. 2009, 48, 7126–7128.
- Phipps, R. J.; Gaunt, M. J. Science 2009, 323, 1593.
- Tremont, S. J.; Rahman, H. U. J. Am. Chem. Soc. 1984, 106, 5759–5760.
- Daugulis, O.; Zaitsev, V. Angew. Chem. Int. Ed. 2005, 44, 4046–4048.
- "Archived copy". Archived from the original on March 4, 2016. Retrieved December 1, 2014.
{{cite web}}: CS1 maint: archived copy as title (link) - Duong, H. A; Gilligan, R. E.; Cooke, M. L.; Phipps, R. J.; Gaunt, M. J. Angew. Chem. Int. Ed. 2011, 50, 463–466.
- Chen, B.; Hou, X-L.; Li, Y-X.; Wu, Y-D. J. Am. Chem. Soc. 2011, 133, 7668–7671.
- Phipps, R. J.; Grimster, N. P.; Gaunt, M. J. J. Am. Chem. Soc. 2008, 130, 8172–8174.
- Maleczka, R. E. Jr. Science 2009, 323, 1572–1573.
- Schwarz, H. Acc. Chem. Res. 1989, 22, 282–287.
- Li, W.; Dastbaravardeh, N.; Li, G.; Yu, J-Q. J. Am. Chem. Soc. 2013, 135, 18056–18059.
- Tang, R-Y.; Li, G.; Yu, J-Q. Nature 2014, 507, 215.
- Cheng, G-J,; Yang, Y-F.; Liu, P.; Chen, P.; Sun, T-Y.; Li, G.; Zhang, X.; Houk, K. N.; Yu, J-Q.; Wu, Y-D. J. Am. Chem. Soc. 2014, 136, 894–897.
- O. Saidi, J. Marafie, A. E. W. Ledger, P. M. Liu, M. F. Mahon, G. Kociok-Kohn, M. K. Whittlesey, and C. G. Frost, J. Am. Chem. Soc., 2011, 133, 19298-19301
- Hofmann, N.; Ackermann, L. J. Am. Chem. Soc. 2013, 135, 5877–5884.
- A. J. Paterson, S. St John-Campbell, M. F. Mahon, N. J. Press, C. G. Frost, Chem. Commun., 2015, 51, 12807-12810.