Bisulfite sequencing
Bisulfite[1] sequencing (also known as bisulphite sequencing) is the use of bisulfite treatment of DNA before routine sequencing to determine the pattern of methylation. DNA methylation was the first discovered epigenetic mark, and remains the most studied. In animals it predominantly involves the addition of a methyl group to the carbon-5 position of cytosine residues of the dinucleotide CpG, and is implicated in repression of transcriptional activity.


Treatment of DNA with bisulfite converts cytosine residues to uracil, but leaves 5-methylcytosine residues unaffected. Therefore, DNA that has been treated with bisulfite retains only methylated cytosines. Thus, bisulfite treatment introduces specific changes in the DNA sequence that depend on the methylation status of individual cytosine residues, yielding single-nucleotide resolution information about the methylation status of a segment of DNA. Various analyses can be performed on the altered sequence to retrieve this information. The objective of this analysis is therefore reduced to differentiating between single nucleotide polymorphisms (cytosines and thymidine) resulting from bisulfite conversion (Figure 1).
Methods
Bisulfite sequencing applies routine sequencing methods on bisulfite-treated genomic DNA to determine methylation status at CpG dinucleotides. Other non-sequencing strategies are also employed to interrogate the methylation at specific loci or at a genome-wide level. All strategies assume that bisulfite-induced conversion of unmethylated cytosines to uracil is complete, and this serves as the basis of all subsequent techniques. Ideally, the method used would determine the methylation status separately for each allele. Alternative methods to bisulfite sequencing include Combined Bisulphite Restriction Analysis and methylated DNA immunoprecipitation (MeDIP).
Methodologies to analyze bisulfite-treated DNA are continuously being developed. To summarize these rapidly evolving methodologies, numerous review articles have been written.[2][3][4][5]
The methodologies can be generally divided into strategies based on methylation-specific PCR (MSP) (Figure 4), and strategies employing polymerase chain reaction (PCR) performed under non-methylation-specific conditions (Figure 3). Microarray-based methods use PCR based on non-methylation-specific conditions also.
Non-methylation-specific PCR based methods
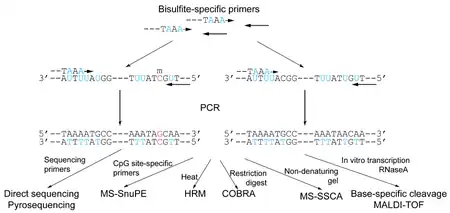
Direct sequencing
The first reported method of methylation analysis using bisulfite-treated DNA utilized PCR and standard dideoxynucleotide DNA sequencing to directly determine the nucleotides resistant to bisulfite conversion.[6] Primers are designed to be strand-specific as well as bisulfite-specific (i.e., primers containing non-CpG cytosines such that they are not complementary to non-bisulfite-treated DNA), flanking (but not involving) the methylation site of interest. Therefore, it will amplify both methylated and unmethylated sequences, in contrast to methylation-specific PCR. All sites of unmethylated cytosines are displayed as thymines in the resulting amplified sequence of the sense strand, and as adenines in the amplified antisense strand. By incorporating high throughput sequencing adaptors into the PCR primers, PCR products can be sequenced with massively parallel sequencing. Alternatively, and labour-intensively, PCR product can be cloned and sequenced. Nested PCR methods can be used to enhance the product for sequencing.
All subsequent DNA methylation analysis techniques using bisulfite-treated DNA is based on this report by Frommer et al. (Figure 2).[6] Although most other modalities are not true sequencing-based techniques, the term "bisulfite sequencing" is often used to describe bisulfite-conversion DNA methylation analysis techniques in general.
Pyrosequencing
Pyrosequencing has also been used to analyze bisulfite-treated DNA without using methylation-specific PCR.[7][8] Following PCR amplification of the region of interest, pyrosequencing is used to determine the bisulfite-converted sequence of specific CpG sites in the region. The ratio of C-to-T at individual sites can be determined quantitatively based on the amount of C and T incorporation during the sequence extension. The main limitation of this method is the cost of the technology. However, Pyrosequencing does well allow for extension to high-throughput screening methods.
A variant of this technique, described by Wong et al., uses allele-specific primers that incorporate single-nucleotide polymorphisms into the sequence of the sequencing primer, thus allowing for separate analysis of maternal and paternal alleles.[9] This technique is of particular usefulness for genomic imprinting analysis.
Methylation-sensitive single-strand conformation analysis (MS-SSCA)
This method is based on the single-strand conformation polymorphism analysis (SSCA) method developed for single-nucleotide polymorphism (SNP) analysis.[10] SSCA differentiates between single-stranded DNA fragments of identical size but distinct sequence based on differential migration in non-denaturating electrophoresis. In MS-SSCA, this is used to distinguish between bisulfite-treated, PCR-amplified regions containing the CpG sites of interest. Although SSCA lacks sensitivity when only a single nucleotide difference is present, bisulfite treatment frequently makes a number of C-to-T conversions in most regions of interest, and the resulting sensitivity approaches 100%. MS-SSCA also provides semi-quantitative analysis of the degree of DNA methylation based on the ratio of band intensities. However, this method is designed to assess all CpG sites as a whole in the region of interest rather than individual methylation sites.
High resolution melting analysis (HRM)
A further method to differentiate converted from unconverted bisulfite-treated DNA is using high-resolution melting analysis (HRM), a quantitative PCR-based technique initially designed to distinguish SNPs.[11] The PCR amplicons are analyzed directly by temperature ramping and resulting liberation of an intercalating fluorescent dye during melting. The degree of methylation, as represented by the C-to-T content in the amplicon, determines the rapidity of melting and consequent release of the dye. This method allows direct quantitation in a single-tube assay, but assesses methylation in the amplified region as a whole rather than at specific CpG sites.
Methylation-sensitive single-nucleotide primer extension (MS-SnuPE)
MS-SnuPE employs the primer extension method initially designed for analyzing single-nucleotide polymorphisms.[12] DNA is bisulfite-converted, and bisulfite-specific primers are annealed to the sequence up to the base pair immediately before the CpG of interest. The primer is allowed to extend one base pair into the C (or T) using DNA polymerase terminating dideoxynucleotides, and the ratio of C to T is determined quantitatively.
A number of methods can be used to determine this C:T ratio. At the beginning, MS-SnuPE relied on radioactive ddNTPs as the reporter of the primer extension. Fluorescence-based methods or Pyrosequencing can also be used.[13] However, matrix-assisted laser desorption ionization/time-of-flight (MALDI-TOF) mass spectrometry analysis to differentiate between the two polymorphic primer extension products can be used, in essence, based on the GOOD assay designed for SNP genotyping. Ion pair reverse-phase high-performance liquid chromatography (IP-RP-HPLC) has also been used to distinguish primer extension products.[14]
Base-specific cleavage/MALDI-TOF
A recently described method by Ehrich et al. further takes advantage of bisulfite-conversions by adding a base-specific cleavage step to enhance the information gained from the nucleotide changes.[15] By first using in vitro transcription of the region of interest into RNA (by adding an RNA polymerase promoter site to the PCR primer in the initial amplification), RNase A can be used to cleave the RNA transcript at base-specific sites. As RNase A cleaves RNA specifically at cytosine and uracil ribonucleotides, base-specificity is achieved by adding incorporating cleavage-resistant dTTP when cytosine-specific (C-specific) cleavage is desired, and incorporating dCTP when uracil-specific (U-specific) cleavage is desired. The cleaved fragments can then be analyzed by MALDI-TOF. Bisulfite treatment results in either introduction/removal of cleavage sites by C-to-U conversions or shift in fragment mass by G-to-A conversions in the amplified reverse strand. C-specific cleavage will cut specifically at all methylated CpG sites. By analyzing the sizes of the resulting fragments, it is possible to determine the specific pattern of DNA methylation of CpG sites within the region, rather than determining the extent of methylation of the region as a whole. This method demonstrated efficacy for high-throughput screening, allowing for interrogation of numerous CpG sites in multiple tissues in a cost-efficient manner.
Methylation-specific PCR (MSP)

This alternative method of methylation analysis also uses bisulfite-treated DNA but avoids the need to sequence the area of interest.[16] Instead, primer pairs are designed themselves to be "methylated-specific" by including sequences complementing only unconverted 5-methylcytosines, or, on the converse, "unmethylated-specific", complementing thymines converted from unmethylated cytosines. Methylation is determined by the ability of the specific primer to achieve amplification. This method is particularly useful to interrogate CpG islands with possibly high methylation density, as increased numbers of CpG pairs in the primer increase the specificity of the assay. Placing the CpG pair at the 3'-end of the primer also improves the sensitivity. The initial report using MSP described sufficient sensitivity to detect methylation of 0.1% of alleles. In general, MSP and its related protocols are considered to be the most sensitive when interrogating the methylation status at a specific locus.
The MethyLight method is based on MSP, but provides a quantitative analysis using quantitative PCR.[17] Methylated-specific primers are used, and a methylated-specific fluorescence reporter probe is also used that anneals to the amplified region. In alternative fashion, the primers or probe can be designed without methylation specificity if discrimination is needed between the CpG pairs within the involved sequences. Quantitation is made in reference to a methylated reference DNA. A modification to this protocol to increase the specificity of the PCR for successfully bisulfite-converted DNA (ConLight-MSP) uses an additional probe to bisulfite-unconverted DNA to quantify this non-specific amplification.[18]
Further methodology using MSP-amplified DNA analyzes the products using melting curve analysis (Mc-MSP).[19] This method amplifies bisulfite-converted DNA with both methylated-specific and unmethylated-specific primers, and determines the quantitative ratio of the two products by comparing the differential peaks generated in a melting curve analysis. A high-resolution melting analysis method that uses both quantitative PCR and melting analysis has been introduced, in particular, for sensitive detection of low-level methylation[20]
Microarray-based methods
Microarray-based methods are a logical extension of the technologies available to analyze bisulfite-treated DNA to allow for genome-wide analysis of methylation.[21] Oligonucleotide microarrays are designed using pairs of oligonucleotide hybridization probes targeting CpG sites of interest. One is complementary to the unaltered methylated sequence, and the other is complementary to the C-to-U-converted unmethylated sequence. The probes are also bisulfite-specific to prevent binding to DNA incompletely converted by bisulfite. The Illumina Methylation Assay is one such assay that applies the bisulfite sequencing technology on a microarray level to generate genome-wide methylation data.
Limitations
5-Hydroxymethylcytosine
Bisulfite sequencing is used widely across mammalian genomes, however complications have arisen with the discovery of a new mammalian DNA modification 5-hydroxymethylcytosine.[22][23] 5-Hydroxymethylcytosine converts to cytosine-5-methylsulfonate upon bisulfite treatment, which then reads as a C when sequenced.[24] Therefore, bisulfite sequencing cannot discriminate between 5-methylcytosine and 5-hydroxymethylcytosine. This means that the output from bisulfite sequencing can no longer be defined as solely DNA methylation, as it is the composite of 5-methylcytosine and 5-hydroxymethylcytosine. The development of Tet-assisted oxidative bisulfite sequencing by Chuan He at the University of Chicago is now able to distinguish between the two modifications at single base resolution.[25]
Incomplete conversion
Bisulfite sequencing relies on the conversion of every single unmethylated cytosine residue to uracil. If conversion is incomplete, the subsequent analysis will incorrectly interpret the unconverted unmethylated cytosines as methylated cytosines, resulting in false positive results for methylation. Only cytosines in single-stranded DNA are susceptible to attack by bisulfite, therefore denaturation of the DNA undergoing analysis is critical.[2] It is important to ensure that reaction parameters such as temperature and salt concentration are suitable to maintain the DNA in a single-stranded conformation and allow for complete conversion. Embedding the DNA in agarose gel has been reported to improve the rate of conversion by keeping strands of DNA physically separate.[26] Incomplete conversion rates can be estimated and adjusted-for after sequencing by including an internal control in the sequencing library, such as lambda phage DNA (which is known to be unmethylated) or by aligning bisulfite sequencing reads to a known unmethylated region in the organism, such as the chloroplast genome.[27]
Degradation of DNA during bisulfite treatment
A major challenge in bisulfite sequencing is the degradation of DNA that takes place concurrently with the conversion. The conditions necessary for complete conversion, such as long incubation times, elevated temperature, and high bisulfite concentration, can lead to the degradation of about 90% of the incubated DNA.[28] Given that the starting amount of DNA is often limited, such extensive degradation can be problematic. The degradation occurs as depurinations resulting in random strand breaks.[29] Therefore, the longer the desired PCR amplicon, the more limited the number of intact template molecules will likely be. This could lead to the failure of the PCR amplification, or the loss of quantitatively accurate information on methylation levels resulting from the limited sampling of template molecules. Thus, it is important to assess the amount of DNA degradation resulting from the reaction conditions employed, and consider how this will affect the desired amplicon. Techniques can also be used to minimize DNA degradation, such as cycling the incubation temperature.[29]
In 2020, New England Biolabs developed NEBNext Enzymatic Methyl-seq an alternative enzymatic approach to minimize DNA damage.[30]
Other concerns
A potentially significant problem following bisulfite treatment is incomplete desulfonation of pyrimidine residues due to inadequate alkalization of the solution. This may inhibit some DNA polymerases, rendering subsequent PCR difficult. However, this situation can be avoided by monitoring the pH of the solution to ensure that desulfonation will be complete.[2]
A final concern is that bisulfite treatment greatly reduces the level of complexity in the sample, which can be problematic if multiple PCR reactions are to be performed (2006).[5] Primer design is more difficult, and inappropriate cross-hybridization is more frequent.
Applications: genome-wide methylation analysis
The advances in bisulfite sequencing have led to the possibility of applying them at a genome-wide scale, where, previously, global measure of DNA methylation was feasible only using other techniques, such as Restriction landmark genomic scanning. The mapping of the human epigenome is seen by many scientists as the logical follow-up to the completion of the Human Genome Project.[31][32] This epigenomic information will be important in understanding how the function of the genetic sequence is implemented and regulated. Since the epigenome is less stable than the genome, it is thought to be important in gene-environment interactions.[33]
Epigenomic mapping is inherently more complex than genome sequencing, however, since the epigenome is much more variable than the genome. One's epigenome varies with age, differs between tissues, is altered by environmental factors, and shows aberrations in diseases. Such rich epigenomic mapping, however, representing different ages, tissue types, and disease states, would yield valuable information on the normal function of epigenetic marks as well as the mechanisms leading to aging and disease.
Direct benefits of epigenomic mapping include probable advances in cloning technology. It is believed that failures to produce cloned animals with normal viability and lifespan result from inappropriate patterns of epigenetic marks. Also, aberrant methylation patterns are well characterized in many cancers. Global hypomethylation results in decreased genomic stability, while local hypermethylation of tumour suppressor gene promoters often accounts for their loss of function. Specific patterns of methylation are indicative of specific cancer types, have prognostic value, and can help to guide the best course of treatment.[32]
Large-scale epigenome mapping efforts are under way around the world and have been organized under the Human Epigenome Project.[33] This is based on a multi-tiered strategy, whereby bisulfite sequencing is used to obtain high-resolution methylation profiles for a limited number of reference epigenomes, while less thorough analysis is performed on a wider spectrum of samples. This approach is intended to maximize the insight gained from a given amount of resources, as high-resolution genome-wide mapping remains a costly undertaking.
Gene-set analysis (for example using tools like DAVID and GoSeq) has been shown to be severely biased when applied to high-throughput methylation data (e.g. genome-wide bisulfite sequencing); it has been suggested that this can be corrected using sample label permutations or using a statistical model to control for differences in the numberes of CpG probes / CpG sites that target each gene.[34]
Oxidative bisulfite sequencing
5-Methylcytosine and 5-hydroxymethylcytosine both read as a C in bisulfite sequencing.[24] Oxidative bisulfite sequencing is a method to discriminate between 5-methylcytosine and 5-hydroxymethylcytosine at single base resolution. The method employs a specific (Tet-assisted) chemical oxidation of 5-hydroxymethylcytosine to 5-formylcytosine, which subsequently converts to uracil during bisulfite treatment.[35] The only base that then reads as a C is 5‑methylcytosine, giving a map of the true methylation status in the DNA sample. Levels of 5‑hydroxymethylcytosine can also be quantified by measuring the difference between bisulfite and oxidative bisulfite sequencing.
References
- Chatterjee A, Stockwell PA, Rodger EJ and Morison IM 2012. Comparison of alignment software for genome-wide bisulphite sequence data. Nucleic Acids Research 40(10): e79.
- Fraga MF, Esteller M (September 2002). "DNA methylation: a profile of methods and applications". BioTechniques. 33 (3): 632, 634, 636–49. doi:10.2144/02333rv01. PMID 12238773.
- El-Maarri O (2003). "Methods: DNA methylation". Peroxisomal Disorders and Regulation of Genes. Advances in Experimental Medicine and Biology. Vol. 544. pp. 197–204. doi:10.1007/978-1-4419-9072-3_23. ISBN 978-0-306-48174-1. PMID 14713229.
- Laird PW (April 2003). "The power and the promise of DNA methylation markers". Nature Reviews. Cancer. 3 (4): 253–66. doi:10.1038/nrc1045. PMID 12671664. S2CID 19574628.
- Callinan PA, Feinberg AP (April 2006). "The emerging science of epigenomics". Human Molecular Genetics. 15 Spec No 1 (90001): R95-101. doi:10.1093/hmg/ddl095. PMID 16651376.
- Frommer M, McDonald LE, Millar DS, Collis CM, Watt F, Grigg GW, et al. (March 1992). "A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands". Proceedings of the National Academy of Sciences of the United States of America. 89 (5): 1827–31. Bibcode:1992PNAS...89.1827F. doi:10.1073/pnas.89.5.1827. PMC 48546. PMID 1542678.
- Colella S, Shen L, Baggerly KA, Issa JP, Krahe R (July 2003). "Sensitive and quantitative universal Pyrosequencing methylation analysis of CpG sites". BioTechniques. 35 (1): 146–50. doi:10.2144/03351md01. PMID 12866414.
- Tost J, Dunker J, Gut IG (July 2003). "Analysis and quantification of multiple methylation variable positions in CpG islands by Pyrosequencing". BioTechniques. 35 (1): 152–6. doi:10.2144/03351md02. PMID 12866415.
- Wong HL, Byun HM, Kwan JM, Campan M, Ingles SA, Laird PW, Yang AS (December 2006). "Rapid and quantitative method of allele-specific DNA methylation analysis". BioTechniques. 41 (6): 734–9. doi:10.2144/000112305. PMID 17191619.
- Bianco T, Hussey D, Dobrovic A (1999). "Methylation-sensitive, single-strand conformation analysis (MS-SSCA): A rapid method to screen for and analyze methylation". Human Mutation. 14 (4): 289–93. doi:10.1002/(SICI)1098-1004(199910)14:4<289::AID-HUMU3>3.0.CO;2-A. PMID 10502775. S2CID 22814772.
- Wojdacz TK, Dobrovic A (2007). "Methylation-sensitive high resolution melting (MS-HRM): a new approach for sensitive and high-throughput assessment of methylation". Nucleic Acids Research. 35 (6): e41. doi:10.1093/nar/gkm013. PMC 1874596. PMID 17289753.
- Gonzalgo ML, Jones PA (June 1997). "Rapid quantitation of methylation differences at specific sites using methylation-sensitive single nucleotide primer extension (Ms-SNuPE)". Nucleic Acids Research. 25 (12): 2529–31. doi:10.1093/nar/25.12.2529. PMC 146734. PMID 9171109.
- Uhlmann K, Brinckmann A, Toliat MR, Ritter H, Nürnberg P (December 2002). "Evaluation of a potential epigenetic biomarker by quantitative methyl-single nucleotide polymorphism analysis". Electrophoresis. 23 (24): 4072–9. doi:10.1002/elps.200290023. PMID 12481262. S2CID 43737807.
- Matin MM, Baumer A, Hornby DP (October 2002). "An analytical method for the detection of methylation differences at specific chromosomal loci using primer extension and ion pair reverse phase HPLC". Human Mutation. 20 (4): 305–11. doi:10.1002/humu.10118. PMID 12325026. S2CID 3178841.
- Ehrich M, Nelson MR, Stanssens P, Zabeau M, Liloglou T, Xinarianos G, et al. (November 2005). "Quantitative high-throughput analysis of DNA methylation patterns by base-specific cleavage and mass spectrometry". Proceedings of the National Academy of Sciences of the United States of America. 102 (44): 15785–90. Bibcode:2005PNAS..10215785E. doi:10.1073/pnas.0507816102. PMC 1276092. PMID 16243968.
- Herman JG, Graff JR, Myöhänen S, Nelkin BD, Baylin SB (September 1996). "Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands". Proceedings of the National Academy of Sciences of the United States of America. 93 (18): 9821–6. Bibcode:1996PNAS...93.9821H. doi:10.1073/pnas.93.18.9821. PMC 38513. PMID 8790415.
- Eads CA, Danenberg KD, Kawakami K, Saltz LB, Blake C, Shibata D, et al. (April 2000). "MethyLight: a high-throughput assay to measure DNA methylation". Nucleic Acids Research. 28 (8): 32e–0. doi:10.1093/nar/28.8.e32. PMC 102836. PMID 10734209.
- Rand K, Qu W, Ho T, Clark SJ, Molloy P (June 2002). "Conversion-specific detection of DNA methylation using real-time polymerase chain reaction (ConLight-MSP) to avoid false positives". Methods. 27 (2): 114–20. doi:10.1016/S1046-2023(02)00062-2. PMID 12095268.
- Akey DT, Akey JM, Zhang K, Jin L (October 2002). "Assaying DNA methylation based on high-throughput melting curve approaches". Genomics. 80 (4): 376–84. doi:10.1006/geno.2002.6851. PMID 12376091.
- Kristensen LS, Mikeska T, Krypuy M, Dobrovic A (April 2008). "Sensitive Melting Analysis after Real Time- Methylation Specific PCR (SMART-MSP): high-throughput and probe-free quantitative DNA methylation detection". Nucleic Acids Research. 36 (7): e42. doi:10.1093/nar/gkn113. PMC 2367707. PMID 18344521.
- Adorján P, Distler J, Lipscher E, Model F, Müller J, Pelet C, et al. (March 2002). "Tumour class prediction and discovery by microarray-based DNA methylation analysis". Nucleic Acids Research. 30 (5): 21e–21. doi:10.1093/nar/30.5.e21. PMC 101257. PMID 11861926.
- Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, BrudnoY, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine inmammalian DNA by MLL partner TET1. Science. 2009;324(5929):930-5.
- Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science.2009;324(5929):929-30.
- Huang Y, Pastor WA, Shen Y, Tahiliani M, Liu DR, Rao A. The Behaviour of 5-Hydroxymethylcytosine in Bisulfite Sequencing. PLOS ONE.2010;5(1):e8888.
- Yu, M., Hon, G. C., Szulwach, K. E., Song, C., Jin, P., Ren, B., He, C. Tet-assisted bisulfite sequencing of 5-hydroxymethylcytosine. Nat. Protocols 2012, 7, 2159.
- Olek A, Oswald J, Walter J (December 1996). "A modified and improved method for bisulphite based cytosine methylation analysis". Nucleic Acids Research. 24 (24): 5064–6. doi:10.1093/nar/24.24.5064. PMC 146326. PMID 9016686.
- Niederhuth, Chad E.; Bewick, Adam J.; Ji, Lexiang; Alabady, Magdy S.; Kim, Kyung Do; Li, Qing; Rohr, Nicholas A.; Rambani, Aditi; Burke, John M.; Udall, Joshua A.; Egesi, Chiedozie; Schmutz, Jeremy; Grimwood, Jane; Jackson, Scott A.; Springer, Nathan M. (2016-09-27). "Widespread natural variation of DNA methylation within angiosperms". Genome Biology. 17 (1): 194. doi:10.1186/s13059-016-1059-0. ISSN 1474-760X.
- Grunau C, Clark SJ, Rosenthal A (July 2001). "Bisulfite genomic sequencing: systematic investigation of critical experimental parameters". Nucleic Acids Research. 29 (13): E65-5. doi:10.1093/nar/29.13.e65. PMC 55789. PMID 11433041.
- Ehrich M, Zoll S, Sur S, van den Boom D (2007). "A new method for accurate assessment of DNA quality after bisulfite treatment". Nucleic Acids Research. 35 (5): e29. doi:10.1093/nar/gkl1134. PMC 1865059. PMID 17259213.
- Gautreau, Isabel (2014-09-29). "NEBNext End Prep Mixture v1". protocols.io. doi:10.17504/protocols.io.cg4tyv. Retrieved 2021-05-19.
- Esteller M (June 2006). "The necessity of a human epigenome project". Carcinogenesis. 27 (6): 1121–5. doi:10.1093/carcin/bgl033. PMID 16699174.
- Bradbury J (December 2003). "Human epigenome project--up and running". PLOS Biology. 1 (3): E82. doi:10.1371/journal.pbio.0000082. PMC 300691. PMID 14691553.
- Jones PA, Martienssen R (December 2005). "A blueprint for a Human Epigenome Project: the AACR Human Epigenome Workshop". Cancer Research. 65 (24): 11241–6. doi:10.1158/0008-5472.CAN-05-3865. PMID 16357125.
- Geeleher P, Hartnett L, Egan LJ, Golden A, Raja Ali RA, Seoighe C (August 2013). "Gene-set analysis is severely biased when applied to genome-wide methylation data". Bioinformatics. 29 (15): 1851–7. doi:10.1093/bioinformatics/btt311. PMID 23732277.
- Booth MJ, Branco MR, Ficz G, Oxley D, Krueger F, Reik W, et al. quantitative sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science. 2012;336(6083):934-7.
External links
- Bisulfite conversion protocol
- Human Epigenome Project (HEP) - Data — by the Sanger Institute
- The Epigenome Network of Excellence