Molecular Operating Environment
Molecular Operating Environment (MOE) is a drug discovery software platform that integrates visualization, modeling and simulations, as well as methodology development, in one package. MOE scientific applications are used by biologists, medicinal chemists and computational chemists in pharmaceutical, biotechnology and academic research. MOE runs on Windows, Linux, Unix, and macOS. Main application areas in MOE include structure-based design,[1] fragment-based design,[2] ligand-based design, pharmacophore discovery, medicinal chemistry applications, biologics applications, structural biology and bioinformatics, protein and antibody modeling, molecular modeling and simulations, virtual screening, cheminformatics & QSAR. The Scientific Vector Language (SVL) is the built-in command, scripting and application development language of MOE.
| Developer(s) | Chemical Computing Group |
|---|---|
| Written in | Scientific Vector Language |
| Operating system | Cross-platform |
| Type | Molecular modelling |
| Website | www |
History
The Molecular Operating Environment was developed by the Chemical Computing Group under the supervision of President/CEO Paul Labute.[3] Founded in 1994[4] and based in Montreal, Quebec, Canada, this private company is dedicated to developing computation software that will challenge, revolutionize, and aid in the scientific methodology. The Chemical Computing Group contains a team of mathematicians, scientists, and software engineers constantly altering and updating MOE in order to improve the fields of theoretical/computational chemistry and biology, molecular modeling, and computer-driven molecular design.[5] Researchers specializing in pharmaceutics (drug-discovery); computational chemistry; biotechnology; bioinformatics; cheminformatics; molecular dynamics, simulations, and modeling are the main clients of the Chemical Computing Group.
Software
As discussed before, MOE is a versatile software with main applications in 3D molecular visualization; structure-based protein-ligand design; antibody and biologics design, structure-based protein engineering; SAR and SPR visualization; ligand-based design; protein, DNA/RNA modeling; virtual screening; 3D pharmacophore screening; fragment-based discovery; structural bioinformatics; molecular mechanics and dynamics; peptide modeling; structural biology; cheminformatics and QSAR.[5]
Molecular Modeling and Simulations
Molecular modeling and simulations is a process often used in computational chemistry, but there is wide application for researchers in a variety of fields. This theoretical approach allows scientists to extensively study the properties of molecules, and using the data can provide insight into how these molecules may behave in biological and/or chemical systems.[6] This information is vital to the design of new materials and chemicals.
Molecular Docking
Molecular docking is a computation study used to primarily analyze the binding affinity of a ligand and a receptor. Often times, proteins are studied using this technique, because data from molecular docking allows scientists to predict if a ligand will bind to a specific molecule and if so, how strongly.[7] Molecular docking can be used to predict the binding mode of already known ligands and/or novel ligands, and as a binding affinity predictive instrument.[8] Binding affinity is measured by the change in energy and the more negative the energy, the more stable the complex and the tighter the ligand binds to the receptor.[9] Data from molecular docking can be used to construct new compounds that are more or less efficient at binding to a specific molecule. Molecular docking is extensively used throughout drug discovery for these reasons.[10]
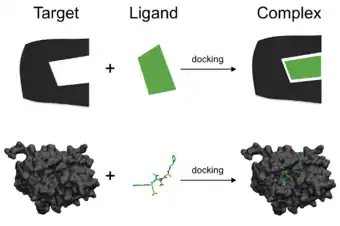
Preparing for molecular docking studies can involve many steps. When docking proteins, proteins are obtained from the Protein Data Bank (PDB), which is an online, open access resources containing the classification, structure/folding, organism, sequence length, mutations, genome, sequence, and other data relating to proteins.[11] The structure of a protein can precisely be determined through a process known as X-ray crystallography. This process involves a concentrated beam of X-rays that is directed at a crystal.[12] When X-rays are projected to a crystal structure, the crystal diffracts the X-rays in specific directions.[13] These directions allow scientists to map and determine the detailed structure of proteins, which is then recorded and uploaded to the PDB.[14]
Methods
The protein structure file is downloaded from the PDB and opened in a molecular docking software. There are many programs that can facilitate molecular docking such as AutoDock, DOCK, FlexX, HYDRO, LIGPLOT, SPROUT, STALK,[15] and Molegro Virtual Docker.[16] Alternatively, some protein structures have not been experimentally determined through the use of X-ray crystallography and therefore, are not found on the PDB. In order to produce a protein molecule that can be used for docking, scientists can use the amino acid sequence of a protein and a program named UniProt to find protein structures in the PDB that have similar amino acid sequences.[17] The amino acid sequence of the protein that is being constructed is then used in combination with the protein structure found in the PDB with the highest percent similarity (template protein) in order to create the target protein used in docking. Although this method does not produce an exact model of the target protein, it allows scientists to produce the closest possible structure in order to conduct computational methods and gain some insight into the behavior of a protein. After constructing the necessary molecules for docking, they are imported into a computational docking software such as MOE. In this program, proteins can be visualized and certain parts of the molecule can be isolated in order to obtain more precise data for a region of interest. A cavity, or region where the molecular docking will take place, is set around the binding site, which is the region in the receptor protein where the ligand attaches to. After specifying the cavity, molecular docking settings are configured and the program is run in order to determine the binding energy of the complex.
Molecular Dynamics (MD)

Molecular dynamic simulations is a computational study that predicts the movement of every atom in a molecule over time.[18] Molecular dynamics can evaluate the movement of water, ions, small and macromolecules, or even complex systems which is extremally useful for reproducing the behavior of chemical and biological environments.[19] This theoretical approach allows scientists to gain further insight into how molecules may behave with respect to each other, specifically if a molecule will leave or remain in a binding pocket. If a molecule remains in a binding pocket, this often indicates that the molecule creates a stable complex with the receptor and is energetically favorable.[20] On the other hand, if the molecule leaves the binding pocket, this indicates that the complex is not stable. This information is then utilized to design new compounds with characteristics that may have a greater or lesser affinity for a receptor.
Applications and Usage
Drug Discovery
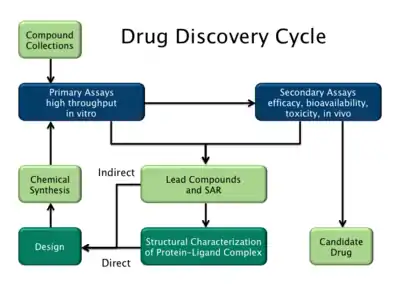
Drug discovery is a process that involves the use of computational, experimental, and clinical studies in order to design new therapeutics.[21] This process is lengthy and costly, yet it is the most popular process to date in developing successful treatments and medicines for a variety of diseases. The increasing use of drug discovery can be attributed to new technology that allow for computation/theoretical studies. Data from computation/theoretical studies is often the foundation and reasoning for the development of new drugs.[22] Without promising theoretical data, these compounds may not be synthesized and tested during experimental studies. Molecular modeling, molecular docking, and MD simulations are some of many computation studies that takes places during drug discovery, allowing scientists to thoroughly study the structure and properties of organic and inorganic molecules. By studying these properties, scientists can gain insight to predict the affinity of molecules in biological and chemical systems in order to determine how a therapeutic may react with different types of chemicals, receptors, and other conditions found in humans or other animals. For example, molecular dynamics is often used throughout drug discovery in order to identify structural cavities that are important for determining binding affinity.[19] This data is then compiled and analyzed to determine if certain therapeutics should be synthesized and tested clinically, or if further optimization is required for the design of new medicines that are more effective.[23]
Pesticides and Herbicides
Computational chemistry can also be applied to the development of safer pesticides and herbicides. Recently, the increasing use of pesticides and herbicides has raised much controversy due to environmental and public health concerns. It was found that although these chemicals are designed to kill target pests, its effects can often harm other organisms, humans included.[24] Some types of pesticides and herbicides such as organophosphates and carbamates can affect the nervous system in humans, while others were found to be carcinogenic, irritate the skin or eyes, and even affect the hormone or endocrine system.[25] Furthermore, neonicotinoids is another type of pesticide that recently gained popularity due to its effectiveness at targeting aphids and other pests that hinder agriculture production.[26] Although there are not many human health concerns associated with neonicotinoids (which is another reason for its popularity), the increasing use of this pesticide has been linked to Colony Collapse Disorder (CCD), or the rapid disappearance of adult bees.[27] Due to this pattern, the European Union has banned the outdoor use of three neonicotinoid pesticides in an attempt to mitigate CCD.[28] Clearly, there are multiple issues regarding the use of these pesticides and herbicides. A call for safer and more efficient pesticides and herbicides is being accomplished with the help of computational/theoretical methods.
Future Implications
Computational/theoretical chemistry and biology methods are continuously pushing the horizon. Recently, DeepMind, which is a company specializing in the development of artificial intelligence (AI), created an AI system named AlphaFold.[29] AlphaFold is the most advanced system to date that can accurately predict a protein's 3D structure from its amino acid sequence.[30] The protein folding problem first began to emerge around the 1960s and ever since, scientists have struggled in determining methods to precisely predict the way a protein will fold solely based on the amino acid sequence.[31] However, with recent advances in technology, AlphaFold has made a breakthrough in this long lasting issue. By utilizing a database with over 350,000 structures, AlphaFold can determine the shape of a protein in a few minutes with atomic accuracy.[32] The ability to predict the structure of millions of unknown proteins can help to combat disease, find more effective medicines, and unlock other unknowns that govern life. This technological breakthrough will revolutionize future research and will have profound effects for the scientific community.
References
- Reynolds CH, Merz KM, Ringe D, eds. (2010). Drug Design: Structure- and Ligand-Based Approaches (1 ed.). Cambridge, UK: Cambridge University Press. ISBN 978-0521887236.
- Erlanson, Daniel A.; McDowell, Robert S.; O'Brien, Tom (2004). "Fragment-Based Drug Discovery". Journal of Medicinal Chemistry. 47 (14): 3463–3482. doi:10.1021/jm040031v. PMID 15214773.
- Frumkin, Daniel; Gayle, Amy Tomlinson; Faraguna, Nico; English, Jen; Sewell, Dawson; Namachunskiy, Rostuslav; Pettitt, Katrina-Kay; Scherfner, Erin; Mešanović, Mevludin. "Chemical Computing Group - Wiki". Golden. Retrieved 2022-12-07.
- "Chemical Computing Group Company Profile: Funding & Investors | PitchBook". pitchbook.com. Retrieved 2022-11-08.
- Chemical Computing Group (November 7, 2022). "Molecular Operating Environment Products". chemcomp.com. Retrieved November 7, 2022.
- "Molecular Simulation - an overview | ScienceDirect Topics". www.sciencedirect.com. Retrieved 2022-11-08.
- Meng, Xuan-Yu; Zhang, Hong-Xing; Mezei, Mihaly; Cui, Meng (2011-06-01). "Molecular Docking: A powerful approach for structure-based drug discovery". Current Computer-Aided Drug Design. 7 (2): 146–157. doi:10.2174/157340911795677602. ISSN 1573-4099. PMC 3151162. PMID 21534921.
- Bartuzi, Damian; Kaczor, Agnieszka A.; Targowska-Duda, Katarzyna M.; Matosiuk, Dariusz (2017-02-22). "Recent Advances and Applications of Molecular Docking to G Protein-Coupled Receptors". Molecules. 22 (2): 340. doi:10.3390/molecules22020340. ISSN 1420-3049. PMC 6155844. PMID 28241450.
- Kastritis, Panagiotis L.; Bonvin, Alexandre M. J. J. (2013-02-06). "On the binding affinity of macromolecular interactions: daring to ask why proteins interact". Journal of the Royal Society Interface. 10 (79): 20120835. doi:10.1098/rsif.2012.0835. PMC 3565702. PMID 23235262.
- Pinzi, Luca; Rastelli, Giulio (2019-09-04). "Molecular Docking: Shifting Paradigms in Drug Discovery". International Journal of Molecular Sciences. 20 (18): 4331. doi:10.3390/ijms20184331. ISSN 1422-0067. PMC 6769923. PMID 31487867.
- Bank, RCSB Protein Data. "RCSB PDB: Homepage". www.rcsb.org. Retrieved 2022-11-08.
- Smyth, M S; Martin, J H J (February 2000). "x Ray crystallography". Molecular Pathology. 53 (1): 8–14. doi:10.1136/mp.53.1.8. ISSN 1366-8714. PMC 1186895. PMID 10884915.
- "X-Ray Crystallography - an overview | ScienceDirect Topics". www.sciencedirect.com. Retrieved 2022-11-08.
- "X-ray Crystallography". Chemistry LibreTexts. 2013-10-02. Retrieved 2022-11-08.
- "6.List of softwares used in molecular docking - Molecular Docking". sites.google.com. Retrieved 2022-11-08.
- Pagadala, Nataraj S.; Syed, Khajamohiddin; Tuszynski, Jack (2017-01-16). "Software for molecular docking: a review". Biophysical Reviews. 9 (2): 91–102. doi:10.1007/s12551-016-0247-1. ISSN 1867-2450. PMC 5425816. PMID 28510083.
- "UniProt". www.uniprot.org. Retrieved 2022-11-08.
- Hollingsworth, Scott A.; Dror, Ron O. (2018-09-19). "Molecular dynamics simulation for all". Neuron. 99 (6): 1129–1143. doi:10.1016/j.neuron.2018.08.011. ISSN 0896-6273. PMC 6209097. PMID 30236283.
- Hernández-Rodríguez, Maricarmen; Rosales-Hernández, Martha C.; Mendieta-Wejebe, Jessica E.; Martínez-Archundia, Marlet; Basurto, José Correa (2016). "Current Tools and Methods in Molecular Dynamics (MD) Simulations for Drug Design". Current Medicinal Chemistry. 23 (34): 3909–3924. doi:10.2174/0929867323666160530144742. ISSN 1875-533X. PMID 27237821.
- "Molecular Dynamics - an overview | ScienceDirect Topics". www.sciencedirect.com. Retrieved 2022-11-09.
- Zhou, Shu-Feng; Zhong, Wei-Zhu (2017-02-13). "Drug Design and Discovery: Principles and Applications". Molecules. 22 (2): 279. doi:10.3390/molecules22020279. ISSN 1420-3049. PMC 6155886. PMID 28208821.
- "Drug Development - an overview | ScienceDirect Topics". www.sciencedirect.com. Retrieved 2022-11-08.
- "Drug discovery - Latest research and news | Nature". www.nature.com. Retrieved 2022-11-08.
- "Pesticides & Human Health | Californians for Pesticide Reform". Retrieved 2022-11-10.
- US EPA, OCSPP (2015-08-11). "Human Health Issues Related to Pesticides". www.epa.gov. Retrieved 2022-11-09.
- May 25; Lindwall, 2022 Courtney. "Neonicotinoids 101: The Effects on Humans and Bees". NRDC. Retrieved 2022-11-09.
- Boston, 677 Huntington Avenue; Ma 02115 +1495‑1000 (2014-05-09). "Study strengthens link between neonicotinoids and collapse of honey bee colonies". News. Retrieved 2022-11-10.
- "European Union expands ban of three neonicotinoid pesticides". www.science.org. Retrieved 2022-11-10.
- "AlphaFold Protein Structure Database". alphafold.ebi.ac.uk. Retrieved 2022-11-10.
- Jumper, John; Evans, Richard; Pritzel, Alexander; Green, Tim; Figurnov, Michael; Ronneberger, Olaf; Tunyasuvunakool, Kathryn; Bates, Russ; Žídek, Augustin; Potapenko, Anna; Bridgland, Alex; Meyer, Clemens; Kohl, Simon A. A.; Ballard, Andrew J.; Cowie, Andrew (August 2021). "Highly accurate protein structure prediction with AlphaFold". Nature. 596 (7873): 583–589. Bibcode:2021Natur.596..583J. doi:10.1038/s41586-021-03819-2. ISSN 1476-4687. PMC 8371605. PMID 34265844.
- Dill, Ken A.; Ozkan, S. Banu; Shell, M. Scott; Weikl, Thomas R. (2008-06-09). "The Protein Folding Problem". Annual Review of Biophysics. 37: 289–316. doi:10.1146/annurev.biophys.37.092707.153558. ISSN 1936-122X. PMC 2443096. PMID 18573083.
- "AlphaFold". www.deepmind.com. Retrieved 2022-11-10.