Molecular lesion
A molecular lesion or point lesion is damage to the structure of a biological molecule such as DNA, RNA, or protein. This damage may result in the reduction or absence of normal function, and in rare cases the gain of a new function. Lesions in DNA may consist of breaks or other changes in chemical structure of the helix, ultimately preventing transcription. Meanwhile, lesions in proteins consist of both broken bonds and improper folding of the amino acid chain. While many nucleic acid lesions are general across DNA and RNA, some are specific to one, such as thymine dimers being found exclusively in DNA. Several cellular repair mechanisms exist, ranging from global to specific, in order to prevent lasting damage resulting from lesions.

Causes
There are two broad causes of nucleic acid lesions, endogenous and exogenous factors. Endogenous factors, or endogeny, refer to the resulting conditions that develop within an organism. This is in contrast with exogenous factors which originate from outside the organism. DNA and RNA lesions caused by endogenous factors generally occur more frequently than damage caused by exogenous ones.[1]
Endogenous Factors
Endogenous sources of specific DNA damage include pathways like hydrolysis, oxidation, alkylation, mismatch of DNA bases, depurination, depyrimidination, double-strand breaks (DSS), and cytosine deamination. DNA lesions can also naturally occur from the release of specific compounds such as reactive oxygen species (ROS), reactive nitrogen species (RNS), reactive carbonyl species (RCS), lipid peroxidation products, adducts, and alkylating agents through metabolic processes. ROS is one of the major endogenous sources of DNA damage and the most studied oxidative DNA adduct is 8-oxo-dG. Other adducts known to form are etheno-, propano-, and malondialdehyde-derived DNA adducts. The aldehydes formed from lipid peroxidation also pose another threat to DNA.[2] Proteins such as “damage-up” proteins (DDPs) can promote endogenous DNA lesions by either increasing the amount of reactive oxygen by transmembrane transporters, losing chromosomes by replisome binding, and stalling replication by transcription factors.[3] For RNA lesions specifically, the most abundant types of endogenous damage include oxidation, alkylation, and chlorination.[4] Phagocytic cells produce radical species that include hypochlorous acid (HOCl), nitric oxide (NO•), and peroxynitrite (ONOO−) to fight infections, and many cell types use nitric oxide as a signaling molecule. However, these radical species can also cause the pathways that form RNA lesions.[5]

Ultraviolet Radiation
UV light, specifically non-ionizing shorter-wavelength radiation such as UVC and UVB, causes direct DNA damage by initiating a synthesis reaction between two thymine molecules. The resulting dimer is very stable. Although they can be removed through excision repairs, when UV damage is extensive, the entire DNA molecule breaks down and the cell dies. If the damage is not too extensive, precancerous or cancerous cells are created from healthy cells.[6]
Chemotherapy drugs
Chemotherapeutics, by design, induce DNA damage and are targeted towards rapidly dividing cancer cells.[7] However, these drugs can not tell the difference between sick and healthy cells, resulting in the damage of normal cells.[8]
Alkylating agents
Alkylating agents are a type of chemotherapeutic drug which keeps the cell from undergoing mitosis by damaging its DNA. They work in all phases of the cell cycle. The use of alkylating agents may result in leukemia due to them being able to target the cells of the bone marrow.[8]
Cancer causing agents
Carcinogens are known to cause a number of DNA lesions, such as single-strand breaks, double- strand breaks, and covalently bound chemical DNA adducts. Tobacco products are one of the most prevalent cancer-causing agents of today.[9] Other DNA damaging, cancer-causing agents include asbestos, which can cause damage through physical interaction with DNA or by indirectly setting off a reactive oxygen species,[10] excessive nickel exposure, which can repress the DNA damage-repair pathways,[11] aflatoxins, which are found in food,[9] and many more.
Lesions of Nucleic Acids

Oxidative lesions
Oxidative lesions are an umbrella category of lesions caused by reactive oxygen species (ROS), reactive nitrogen species (RNS), other byproducts of cellular metabolism, and exogenous factors such as ionizing or ultraviolet radiation.[12] Byproducts of oxidative respiration are the main source of reactive species which cause a background level of oxidative lesions in the cell. DNA and RNA are both affected by this, and it has been found that RNA oxidative lesions are more abundant in humans compared to DNA. This may be due cytoplasmic RNA having closer proximity to the electron transport chain.[13] Known oxidative lesions characterized in DNA and RNA are many in number, as oxidized products are unstable and may resolve quickly. The hydroxyl radical and singlet oxygen are common reactive oxygen species responsible for these lesions.[14] 8-oxo-guanine (8-oxoG) is the most abundant and well characterized oxidative lesion, found in both RNA and DNA. Accumulation of 8-oxoG may cause dire damage within the mitochondria and is thought to be a key player in the aging process.[15] RNA oxidation has direct consequences in the production of proteins. mRNA affected by oxidative lesions is still recognized by ribosome, but the ribosome will undergo stalling and dysfunction. This results in proteins having either decreased expression or truncation, leading to aggregation and general dysfunction.[16]
Structural rearrangements
- Depurination is caused by hydrolysis and results in loss if the purine base of a nucleic acid. DNA is more prone to this, as the transition state in the depurination reaction has much greater energy in RNA.[17]
- Tautomerization is a chemical reaction that is primarily relevant in the behavior of amino acids and nucleic acids. Both of which are correlated to DNA and RNA. The process of tautomerization of DNA bases occurs during DNA replication. The ability for the wrong tautomer of one of the standard nucleic bases to mispair causes a mutation during the process of DNA replication which can be cytotoxic or mutagenic to the cell. These mispairings can result in transition, transversion, frameshift, deletion, and/or duplication mutations.[18] Some diseases that result from tautomerization induced DNA lesions include Kearns-Sayre syndrome, Fragile X syndrome, Kennedy disease, and Huntington’s disease.[18]
- Cytosine deamination commonly occurs under physiological conditions and essentially is the deamination of cytosine. This process yields uracil as its product, which is not a base pair found within DNA. This process causes extensive DNA damage. The rate of this process is slowed down significantly in double-stranded DNA compared to single-stranded DNA.[19]
Single and Double Stranded Breaks
Single-strand breaks (SSBs) occur when one strand of the DNA double helix experiences breakage of a single nucleotide accompanied by damaged 5’- and/or 3’-termini at this point. One common source of SSBs is due to oxidative attack by physiological reactive oxygen species (ROS) such as hydrogen peroxide. H2O2 causes SSBs three times more frequently than double-strand breaks (DSBs). Alternative methods of SSB acquisition include direct disintegration of the oxidized sugar or through DNA base-excision repair (BER) of damaged bases. Additionally, cellular enzymes may perform erroneous activity leading to SSBs or DSBs by a variety of mechanisms. One such example would be when the cleavage complex formed by DNA topoisomerase 1 (TOP1) relaxes DNA during transcription and replication through the transient formation of a nick. While TOP1 normally reseals this nick shortly after, these cleavage complexes may collide with RNA or DNA polymerases or be proximal to other lesions, leading to TOP1-linked SSBs or TOP1-linked DSBs.[20]
Chemical Adducts
A DNA adduct is a segment of DNA that binds to a chemical carcinogen. Some adducts that cause lesions to DNA included oxidatively modified bases, propano-, etheno-, and MDA-induced adducts.[2] 5‐Hydroxymethyluracil is an example of an oxidatively modified base where oxidation of the methyl group of thymine occurs.[21] This adduct interferes with the binding of transcription factors to DNA which can trigger apoptosis or result in deletion mutations.[21] Propano adducts are derived by species generated by lipid peroxidation. For example, HNE is a major toxic product of the process.[22] It regulates the expression of genes that are involved in cell cycle regulation and apoptosis. Some of the aldehydes from lipid peroxidation can be converted to epoxy aldehydes by oxidation reactions.[23] These epoxy aldehydes can damage DNA by producing etheno adducts. An increase in this type of DNA lesion exhibits conditions resulting in oxidative stress which is known to be associated with an increased risk of cancer.[24] Malondialdehyde (MDA) is another highly toxic product from lipid peroxidation and also in the synthesis of prostaglandin. MDA reacts with DNA to form the M1dG adduct which causes DNA lesions.[2]
Disease Effects
Many systems are in place to repair DNA and RNA lesions but it is possible for lesions to escape these measures. This may lead to mutations or large genome abnormalities, which can threaten the cell or organism’s ability to live. Several cancers are a result of DNA lesions. Even repair mechanisms to heal the damage may end up causing more damage. Mismatch repair defects, for example, cause instability that predisposes to colorectal and endometrial carcinomas.[9]
DNA lesions in neurons may lead to neurodegenerative disorders such as Alzheimer’s, Huntington’s, and Parkinson’s diseases. These come as a result of neurons generally being associated with high mitochondrial respiration and redox species production, which can damage nuclear DNA. Since these cells often cannot be replaced after being damaged, the damage done to them leads to dire consequences. Other disorders stemming from DNA lesions and their association with neurons include but are not limited to Fragile X syndrome, Friedrich’s ataxia, and spinocerebellar ataxias.[9]
During replication, usually DNA polymerases are unable to go past the lesioned area, however, some cells are equipped with special polymerases which allow for translesion synthesis (TLS). TLS polymerases allow for the replication of DNA past lesions and risk generating mutations at a high frequency. Common mutations that occur after undergoing this process are point mutations and frameshift mutations. Several diseases come as a result of this process including several cancers and Xeroderma pigmentosum.[25]
The effect of oxidatively damaged RNA has resulted in a number of human diseases and is especially associated with chronic degeneration. This type of damage has been observed in many neurodegenerative diseases such as Amyotrophic lateral sclerosis,[9] Alzheimer’s, Parkinson’s, dementia with Lewy bodies, and several prion diseases.[26] It is important to note that this list is rapidly growing and data suggests that RNA oxidation occurs early in the development of these diseases, rather than as an effect of cellular decay.[9] RNA and DNA lesions are both associated with the development of diabetes mellitus type 2.[9]
Repair Mechanisms
DNA Damage Response
When DNA is damaged such as due to a lesion, a complex signal transduction pathway is activated which is responsible for recognizing the damage and instigating the cell’s response for repair. Compared to the other lesion repair mechanisms, DDR is the highest level of repair and is employed for the most complex lesions. DDR consists of various pathways, the most common of which are the DDR kinase signaling cascades. These are controlled by phosphatidylinositol 3-kinase-related kinases (PIKK), and range from DNA-dependent protein kinase (DNA-PKcs) and ataxia telangiectasia-mutated (ATM) most involved in repairing DSBs to the more versatile Rad3-related (ATR). ATR is crucial to human cell viability, while ATM mutations cause the severe disorder ataxia-telangiectasia leading to neurodegeneration, cancer, and immunodeficiency. These three DDR kinases all recognize damage via protein-protein interactions which localize the kinases to the areas of damage. Next, further protein-protein interactions and posttranslational modifications (PTMs) complete the kinase activation, and a series of phosphorylation events takes place. DDR kinases perform repair regulation at three levels - via PTMs, at the level of chromatin, and at the level of the nucleus.[27]
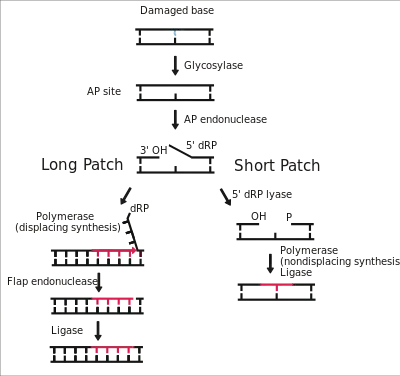
Base Excision Repair
Base excision repair (BER) is responsible for removing damaged bases in DNA. This mechanism specifically works on excising small base lesions which do not distort the DNA double helix, in contrast to the nucleotide excision repair pathway which is employed in correcting more prominent distorting lesions. DNA glycosylases initiate BER by both recognizing the faulty or incorrect bases and then removing them, forming AP sites lacking any purine or pyrimidine. AP endonuclease then cleaves the AP site, and the single-strand break is either processed by short-patch BER to replace a single nucleotide long-patch BER to create 2-10 replacement nucleotides.[28]
Single Stranded Break Repair
Single stranded breaks (SSBs) can severely threaten genetic stability and cell survival if not quickly and properly repaired, so cells have developed fast and efficient SSB repair (SSBR) mechanisms. While global SSBR systems extract SSBs throughout the genome and during interphase, S-phase specific SSBR processes work together with homologous recombination at the replication forks.[29]
Double Stranded Break Repair
Double stranded breaks (DSB) are a threat to all organisms as they can cause cell death and cancer. They can be caused exogenously as a result of radiation and endogenously from errors in replication or encounters with DNA lesions by the replication fork.[30] DSB repair occurs through a variety of different pathways and mechanisms in order to correctly repair these errors.
Nucleotide Excision and Mismatch Repair
Nucleotide excision repair is one of the main mechanisms used to remove bulky adducts from DNA lesions caused by chemotherapy drugs, environmental mutagens, and most importantly UV radiation.[9] This mechanism functions by releasing a short damage containing oligonucleotide from the DNA site, and then that gap is filled in and repaired by NER.[9] NER recognizes a variety of structurally unrelated DNA lesions due to the flexibility of the mechanism itself, as NER is highly sensitive to changes in the DNA helical structure.[31] Bulky adducts seem to trigger NER.[31] The XPC-RAD23-CETN2 heterotrimer involved with NER has a critical role in DNA lesion recognition.[32] In addition to other general lesions in the genome, UV damaged DNA binding protein complex (UV-DDB) also has an important role in both recognition and repair of UV-induced DNA photolesions.[32]
Mismatch repair (MMR) mechanisms within the cell correct base mispairs that occur during replication using a variety of pathways. It has a high affinity for targeting DNA lesions with specificity, as alternations in base pair stacking that occur at DNA lesion sites affect the helical structure.[33] This is likely one of many signals that triggers MMR.
References
- Chakarov, Stoyan; Petkova, Rumena; Russev, George Ch; Zhelev, Nikolai (2014-02-23). "DNA damage and mutation. Types of DNA damage". BioDiscovery. 11 (11): e8957. Bibcode:2014BioDi..11....1C. doi:10.7750/BioDiscovery.2014.11.1. ISSN 2050-2966.
- De Bont, Rinne; van Larebeke, Nik (2004-05-01). "Endogenous DNA damage in humans: a review of quantitative data". Mutagenesis. 19 (3): 169–185. doi:10.1093/mutage/geh025. ISSN 0267-8357. PMID 15123782.
- Xia, Jun; Chiu, Li-Ya; Nehring, Ralf B.; Núñez, María Angélica Bravo; Mei, Qian; Perez, Mercedes; Zhai, Yin; Fitzgerald, Devon M.; Pribis, John P.; Wang, Yumeng; Hu, Chenyue W. (2019-01-10). "Bacteria-to-human protein networks reveal origins of endogenous DNA damage". Cell. 176 (1–2): 127–143.e24. doi:10.1016/j.cell.2018.12.008. ISSN 0092-8674. PMC 6344048. PMID 30633903.
- Yan, Liewei L.; Zaher, Hani S. (2019-10-11). "How do cells cope with RNA damage and its consequences?". The Journal of Biological Chemistry. 294 (41): 15158–15171. doi:10.1074/jbc.REV119.006513. ISSN 0021-9258. PMC 6791314. PMID 31439666.
- Wurtmann, Elisabeth J.; Wolin, Sandra L. (2009). "RNA under attack: Cellular handling of RNA damage". Critical Reviews in Biochemistry and Molecular Biology. 44 (1): 34–49. doi:10.1080/10409230802594043. ISSN 1040-9238. PMC 2656420. PMID 19089684.
- "How does ultraviolet light kill cells?". Scientific American. Retrieved 2020-11-30.
- Woods, Derek; Turchi, John J. (2013-05-01). "Chemotherapy induced DNA damage response". Cancer Biology & Therapy. 14 (5): 379–389. doi:10.4161/cbt.23761. ISSN 1538-4047. PMC 3672181. PMID 23380594.
- "How Chemotherapy Drugs Work". www.cancer.org. Retrieved 2020-11-30.
- Jackson, Stephen P.; Bartek, Jiri (2009-10-22). "The DNA-damage response in human biology and disease". Nature. 461 (7267): 1071–1078. Bibcode:2009Natur.461.1071J. doi:10.1038/nature08467. ISSN 0028-0836. PMC 2906700. PMID 19847258.
- Okayasu, Ryuichi; Takahashi, Sentaro; Yamada, Shigeru; Hei, Tom K.; Ullrich, Robert L. (1999-01-15). "Asbestos and DNA Double Strand Breaks". Cancer Research. 59 (2): 298–300. ISSN 0008-5472. PMID 9927035.
- Guo, Hongrui; Liu, Huan; Wu, Hongbin; Cui, Hengmin; Fang, Jing; Zuo, Zhicai; Deng, Junliang; Li, Yinglun; Wang, Xun; Zhao, Ling (2019-09-21). "Nickel Carcinogenesis Mechanism: DNA Damage". International Journal of Molecular Sciences. 20 (19): 4690. doi:10.3390/ijms20194690. ISSN 1422-0067. PMC 6802009. PMID 31546657.
- Cooke, Marcus S.; Evans, Mark D.; Dizdaroglu, Miral; Lunec, Joseph (2003). "Oxidative DNA damage: mechanisms, mutation, and disease". The FASEB Journal. 17 (10): 1195–1214. doi:10.1096/fj.02-0752rev. ISSN 1530-6860. PMID 12832285. S2CID 1132537.
- Weimann, Allan; Belling, Dorthe; Poulsen, Henrik E. (2002-01-15). "Quantification of 8-oxo-guanine and guanine as the nucleobase, nucleoside and deoxynucleoside forms in human urine by high-performance liquid chromatography–electrospray tandem mass spectrometry". Nucleic Acids Research. 30 (2): e7. doi:10.1093/nar/30.2.e7. ISSN 0305-1048. PMC 99846. PMID 11788733.
- Calabretta, Alessandro; Küpfer, Pascal A.; Leumann, Christian J. (2015-05-19). "The effect of RNA base lesions on mRNA translation". Nucleic Acids Research. 43 (9): 4713–4720. doi:10.1093/nar/gkv377. ISSN 1362-4962. PMC 4482091. PMID 25897124.
- Radak, Zsolt; Boldogh, Istvan (2010-08-15). "8-oxo-7,8-dihydroguanine: Link to gene expression, aging and defense against oxidative stress". Free Radical Biology & Medicine. 49 (4): 587–596. doi:10.1016/j.freeradbiomed.2010.05.008. ISSN 0891-5849. PMC 2943936. PMID 20483371.
- Poulsen, Henrik E.; Specht, Elisabeth; Broedbaek, Kasper; Henriksen, Trine; Ellervik, Christina; Mandrup-Poulsen, Thomas; Tonnesen, Morten; Nielsen, Peter E.; Andersen, Henrik U.; Weimann, Allan (2012-04-15). "RNA modifications by oxidation: a novel disease mechanism?". Free Radical Biology & Medicine. 52 (8): 1353–1361. doi:10.1016/j.freeradbiomed.2012.01.009. ISSN 1873-4596. PMID 22306201.
- Cavalieri, Ercole; Saeed, Muhammad; Zahid, Muhammad; Cassada, David; Snow, Daniel; Miljkovic, Momcilo; Rogan, Eleanor (February 2012). "Mechanism of DNA Depurination by Carcinogens in Relation to Cancer Initiation". IUBMB Life. 64 (2): 169–179. doi:10.1002/iub.586. ISSN 1521-6543. PMC 4418633. PMID 22162200.
- Griffiths, Anthony JF; Miller, Jeffrey H.; Suzuki, David T.; Lewontin, Richard C.; Gelbart, William M. (2000). "Spontaneous mutations". An Introduction to Genetic Analysis. 7th Edition.
- "DNA Damage - the major cause of missing pieces from the DNA puzzle | NEB". www.neb.com. Retrieved 2020-12-01.
- Caldecott, Keith W. (August 2008). "Single-strand break repair and genetic disease". Nature Reviews Genetics. 9 (8): 619–631. doi:10.1038/nrg2380. ISSN 1471-0064. PMID 18626472. S2CID 39412478.
- Rogstad, Daniel K.; Liu, Pingfang; Burdzy, Artur; Lin, Susan S.; Sowers, Lawrence C. (2002-06-25). "Endogenous DNA lesions can inhibit the binding of the AP-1 (c-Jun) transcription factor". Biochemistry. 41 (25): 8093–8102. doi:10.1021/bi012180a. ISSN 0006-2960. PMID 12069602.
- Esterbauer, H.; Eckl, P.; Ortner, A. (May 1990). "Possible mutagens derived from lipids and lipid precursors". Mutation Research. 238 (3): 223–233. doi:10.1016/0165-1110(90)90014-3. ISSN 0027-5107. PMID 2342513.
- Chung, F. L.; Chen, H. J.; Nath, R. G. (October 1996). "Lipid peroxidation as a potential endogenous source for the formation of exocyclic DNA adducts". Carcinogenesis. 17 (10): 2105–2111. doi:10.1093/carcin/17.10.2105. ISSN 0143-3334. PMID 8895475.
- Bartsch, H.; Nair, J. (2000-11-16). "Ultrasensitive and specific detection methods for exocylic DNA adducts: markers for lipid peroxidation and oxidative stress". Toxicology. 153 (1–3): 105–114. doi:10.1016/s0300-483x(00)00307-3. ISSN 0300-483X. PMID 11090950.
- Pagès, Vincent; Fuchs, Robert PP (December 2002). "How DNA lesions are turned into mutations within cells?". Oncogene. 21 (58): 8957–8966. doi:10.1038/sj.onc.1206006. ISSN 1476-5594. PMID 12483512. S2CID 25226350.
- Fimognari, Carmela (2015). "Role of Oxidative RNA Damage in Chronic-Degenerative Diseases". Oxidative Medicine and Cellular Longevity. 2015: 358713. doi:10.1155/2015/358713. ISSN 1942-0900. PMC 4452857. PMID 26078805.
- Sirbu, Bianca M.; Cortez, David (August 2013). "DNA Damage Response: Three Levels of DNA Repair Regulation". Cold Spring Harbor Perspectives in Biology. 5 (8): a012724. doi:10.1101/cshperspect.a012724. ISSN 1943-0264. PMC 3721278. PMID 23813586.
- Krokan, Hans E.; Bjørås, Magnar (April 2013). "Base Excision Repair". Cold Spring Harbor Perspectives in Biology. 5 (4): a012583. doi:10.1101/cshperspect.a012583. ISSN 1943-0264. PMC 3683898. PMID 23545420.
- Jackson, Stephen P. (2002-05-01). "Sensing and repairing DNA double-strand breaks". Carcinogenesis. 23 (5): 687–696. doi:10.1093/carcin/23.5.687. ISSN 0143-3334. PMID 12016139.
- Cannan, Wendy J.; Pederson, David S. (January 2016). "Mechanisms and Consequences of Double-strand DNA Break Formation in Chromatin". Journal of Cellular Physiology. 231 (1): 3–14. doi:10.1002/jcp.25048. ISSN 0021-9541. PMC 4994891. PMID 26040249.
- de Boer, Jan; Hoeijmakers, Jan H. J. (2000-03-01). "Nucleotide excision repair and human syndromes". Carcinogenesis. 21 (3): 453–460. doi:10.1093/carcin/21.3.453. ISSN 0143-3334. PMID 10688865.
- Kusakabe, Masayuki; Onishi, Yuki; Tada, Haruto; Kurihara, Fumika; Kusao, Kanako; Furukawa, Mari; Iwai, Shigenori; Yokoi, Masayuki; Sakai, Wataru; Sugasawa, Kaoru (2019-01-25). "Mechanism and regulation of DNA damage recognition in nucleotide excision repair". Genes and Environment. 41 (1): 2. doi:10.1186/s41021-019-0119-6. ISSN 1880-7062. PMC 6346561. PMID 30700997.
- Hsieh, Peggy; Yamane, Kazuhiko (2008). "DNA mismatch repair: Molecular mechanism, cancer, and ageing". Mechanisms of Ageing and Development. 129 (7–8): 391–407. doi:10.1016/j.mad.2008.02.012. ISSN 0047-6374. PMC 2574955. PMID 18406444.