Nitrogen compounds
The chemical element nitrogen is one of the most abundant elements in the universe and can form many compounds. It can take several oxidation states; but the most common oxidation states are -3 and +3. Nitrogen can form nitride and nitrate ions. It also forms a part of nitric acid and nitrate salts. Nitrogen compounds also have an important role in organic chemistry, as nitrogen is part of proteins, amino acids and adenosine triphosphate.
Dinitrogen complexes
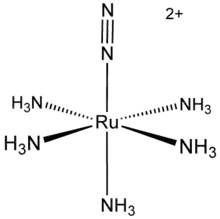
The first example of a dinitrogen complex to be discovered was [Ru(NH3)5(N2)]2+ (see figure at right), and soon many other such complexes were discovered. These complexes, in which a nitrogen molecule donates at least one lone pair of electrons to a central metal cation, illustrate how N2 might bind to the metal(s) in nitrogenase and the catalyst for the Haber process: these processes involving dinitrogen activation are vitally important in biology and in the production of fertilisers.[1][2]
Dinitrogen is able to coordinate to metals in five different ways. The more well-characterised ways are the end-on M←N≡N (η1) and M←N≡N→M (μ, bis-η1), in which the lone pairs on the nitrogen atoms are donated to the metal cation. The less well-characterised ways involve dinitrogen donating electron pairs from the triple bond, either as a bridging ligand to two metal cations (μ, bis-η2) or to just one (η2). The fifth and unique method involves triple-coordination as a bridging ligand, donating all three electron pairs from the triple bond (μ3-N2). A few complexes feature multiple N2 ligands and some feature N2 bonded in multiple ways. Since N2 is isoelectronic with carbon monoxide (CO) and acetylene (C2H2), the bonding in dinitrogen complexes is closely allied to that in carbonyl compounds, although N2 is a weaker σ-donor and π-acceptor than CO. Theoretical studies show that σ donation is a more important factor allowing the formation of the M–N bond than π back-donation, which mostly only weakens the N–N bond, and end-on (η1) donation is more readily accomplished than side-on (η2) donation.[3]
Today, dinitrogen complexes are known for almost all the transition metals, accounting for several hundred compounds. They are normally prepared by three methods:[3]
- Replacing labile ligands such as H2O, H−, or CO directly by nitrogen: these are often reversible reactions that proceed at mild conditions.
- Reducing metal complexes in the presence of a suitable coligand in excess under nitrogen gas. A common choice include replacing chloride ligands by dimethylphenylphosphine (PMe2Ph) to make up for the smaller number of nitrogen ligands attached than the original chlorine ligands.
- Converting a ligand with N–N bonds, such as hydrazine or azide, directly into a dinitrogen ligand.
Occasionally the N≡N bond may be formed directly within a metal complex, for example by directly reacting coordinated ammonia (NH3) with nitrous acid (HNO2), but this is not generally applicable. Most dinitrogen complexes have colours within the range white-yellow-orange-red-brown; a few exceptions are known, such as the blue [{Ti(η5-C5H5)2}2-(N2)].[3]
Nitrides, azides, and nitrido complexes
Nitrogen bonds to almost all the elements in the periodic table except the first three noble gases, helium, neon, and argon, and some of the very short-lived elements after bismuth, creating an immense variety of binary compounds with varying properties and applications.[3] Many binary compounds are known: with the exception of the nitrogen hydrides, oxides, and fluorides, these are typically called nitrides. Many stoichiometric phases are usually present for most elements (e.g. MnN, Mn6N5, Mn3N2, Mn2N, Mn4N, and MnxN for 9.2 < x < 25.3). They may be classified as "salt-like" (mostly ionic), covalent, "diamond-like", and metallic (or interstitial), although this classification has limitations generally stemming from the continuity of bonding types instead of the discrete and separate types that it implies. They are normally prepared by directly reacting a metal with nitrogen or ammonia (sometimes after heating), or by thermal decomposition of metal amides:[4]
- 3 Ca + N2 → Ca3N2
- 3 Mg + 2 NH3 → Mg3N2 + 3 H2 (at 900 °C)
- 3 Zn(NH2)2 → Zn3N2 + 4 NH3
Many variants on these processes are possible. The most ionic of these nitrides are those of the alkali metals and alkaline earth metals, Li3N (Na, K, Rb, and Cs do not form stable nitrides for steric reasons) and M3N2 (M = Be, Mg, Ca, Sr, Ba). These can formally be thought of as salts of the N3− anion, although charge separation is not actually complete even for these highly electropositive elements. However, the alkali metal azides NaN3 and KN3, featuring the linear N−
3 anion, are well-known, as are Sr(N3)2 and Ba(N3)2. Azides of the B-subgroup metals (those in groups 11 through 16) are much less ionic, have more complicated structures, and detonate readily when shocked.[4]
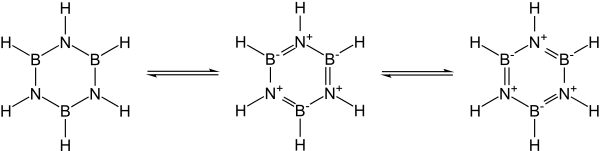
Many covalent binary nitrides are known. Examples include cyanogen ((CN)2), triphosphorus pentanitride (P3N5), disulfur dinitride (S2N2), and tetrasulfur tetranitride (S4N4). The essentially covalent silicon nitride (Si3N4) and germanium nitride (Ge3N4) are also known: silicon nitride in particular would make a promising ceramic if not for the difficulty of working with and sintering it. In particular, the group 13 nitrides, most of which are promising semiconductors, are isoelectronic with graphite, diamond, and silicon carbide and have similar structures: their bonding changes from covalent to partially ionic to metallic as the group is descended. In particular, since the B–N unit is isoelectronic to C–C, and carbon is essentially intermediate in size between boron and nitrogen, much of organic chemistry finds an echo in boron–nitrogen chemistry, such as in borazine ("inorganic benzene"). Nevertheless, the analogy is not exact due to the ease of nucleophilic attack at boron due to its deficiency in electrons, which is not possible in a wholly carbon-containing ring.[4]
The largest category of nitrides are the interstitial nitrides of formulae MN, M2N, and M4N (although variable composition is perfectly possible), where the small nitrogen atoms are positioned in the gaps in a metallic cubic or hexagonal close-packed lattice. They are opaque, very hard, and chemically inert, melting only at very high temperatures (generally over 2500 °C). They have a metallic lustre and conduct electricity as do metals. They hydrolyse only very slowly to give ammonia or nitrogen.[4]
The nitride anion (N3−) is the strongest π donor known amongst ligands (the second-strongest is O2−). Nitrido complexes are generally made by thermal decomposition of azides or by deprotonating ammonia, and they usually involve a terminal {≡N}3− group. The linear azide anion (N−
3), being isoelectronic with nitrous oxide, carbon dioxide, and cyanate, forms many coordination complexes. Further catenation is rare, although N4−
4 (isoelectronic with carbonate and nitrate) is known.[4]
Hydrides

Industrially, ammonia (NH3) is the most important compound of nitrogen and is prepared in larger amounts than any other compound, because it contributes significantly to the nutritional needs of terrestrial organisms by serving as a precursor to food and fertilisers. It is a colourless alkaline gas with a characteristic pungent smell. The presence of hydrogen bonding has very significant effects on ammonia, conferring on it its high melting (−78 °C) and boiling (−33 °C) points. As a liquid, it is a very good solvent with a high heat of vaporisation (enabling it to be used in vacuum flasks), that also has a low viscosity and electrical conductivity and high dielectric constant, and is less dense than water. However, the hydrogen bonding in NH3 is weaker than that in H2O due to the lower electronegativity of nitrogen compared to oxygen and the presence of only one lone pair in NH3 rather than two in H2O. It is a weak base in aqueous solution (pKb 4.74); its conjugate acid is ammonium, NH+
4. It can also act as an extremely weak acid, losing a proton to produce the amide anion, NH−
2. It thus undergoes self-dissociation, similar to water, to produce ammonium and amide. Ammonia burns in air or oxygen, though not readily, to produce nitrogen gas; it burns in fluorine with a greenish-yellow flame to give nitrogen trifluoride. Reactions with the other nonmetals are very complex and tend to lead to a mixture of products. Ammonia reacts on heating with metals to give nitrides.[6]
Many other binary nitrogen hydrides are known, but the most important are hydrazine (N2H4) and hydrogen azide (HN3). Although it is not a nitrogen hydride, hydroxylamine (NH2OH) is similar in properties and structure to ammonia and hydrazine as well. Hydrazine is a fuming, colourless liquid that smells similarly to ammonia. Its physical properties are very similar to those of water (melting point 2.0 °C, boiling point 113.5 °C, density 1.00 g/cm3). Despite it being an endothermic compound, it is kinetically stable. It burns quickly and completely in air very exothermically to give nitrogen and water vapour. It is a very useful and versatile reducing agent and is a weaker base than ammonia.[7] It is also commonly used as a rocket fuel.[8]
Hydrazine is generally made by reaction of ammonia with alkaline sodium hypochlorite in the presence of gelatin or glue:[7]
- NH3 + OCl− → NH2Cl + OH−
- NH2Cl + NH3 → N
2H+
5 + Cl− (slow) - N
2H+
5 + OH− → N2H4 + H2O (fast)
(The attacks by hydroxide and ammonia may be reversed, thus passing through the intermediate NHCl− instead.) The reason for adding gelatin is that it removes metal ions such as Cu2+ that catalyses the destruction of hydrazine by reaction with monochloramine (NH2Cl) to produce ammonium chloride and nitrogen.[7]
Hydrogen azide (HN3) was first produced in 1890 by the oxidation of aqueous hydrazine by nitrous acid. It is very explosive and even dilute solutions can be dangerous. It has a disagreeable and irritating smell and is a potentially lethal (but not cumulative) poison. It may be considered the conjugate acid of the azide anion, and is similarly analogous to the hydrohalic acids.[7]
Halides and oxohalides

All four simple nitrogen trihalides are known. A few mixed halides and hydrohalides are known, but are mostly unstable; examples include NClF2, NCl2F, NBrF2, NF2H, NFH2, NCl2H, and NClH2.[9]
Five nitrogen fluorides are known. Nitrogen trifluoride (NF3, first prepared in 1928) is a colourless and odourless gas that is thermodynamically stable, and most readily produced by the electrolysis of molten ammonium fluoride dissolved in anhydrous hydrogen fluoride. Like carbon tetrafluoride, it is not at all reactive and is stable in water or dilute aqueous acids or alkalis. Only when heated does it act as a fluorinating agent, and it reacts with copper, arsenic, antimony, and bismuth on contact at high temperatures to give tetrafluorohydrazine (N2F4). The cations NF+
4 and N
2F+
3 are also known (the latter from reacting tetrafluorohydrazine with strong fluoride-acceptors such as arsenic pentafluoride), as is ONF3, which has aroused interest due to the short N–O distance implying partial double bonding and the highly polar and long N–F bond. Tetrafluorohydrazine, unlike hydrazine itself, can dissociate at room temperature and above to give the radical NF2•. Fluorine azide (FN3) is very explosive and thermally unstable. Dinitrogen difluoride (N2F2) exists as thermally interconvertible cis and trans isomers, and was first found as a product of the thermal decomposition of FN3.[9]
Nitrogen trichloride (NCl3) is a dense, volatile, and explosive liquid whose physical properties are similar to those of carbon tetrachloride, although one difference is that NCl3 is easily hydrolysed by water while CCl4 is not. It was first synthesised in 1811 by Pierre Louis Dulong, who lost three fingers and an eye to its explosive tendencies. As a dilute gas it is less dangerous and is thus used industrially to bleach and sterilise flour. Nitrogen tribromide (NBr3), first prepared in 1975, is a deep red, temperature-sensitive, volatile solid that is explosive even at −100 °C. Nitrogen triiodide (NI3) is still more unstable and was only prepared in 1990. Its adduct with ammonia, which was known earlier, is very shock-sensitive: it can be set off by the touch of a feather, shifting air currents, or even alpha particles.[9][10] For this reason, small amounts of nitrogen triiodide are sometimes synthesised as a demonstration to high school chemistry students or as an act of "chemical magic".[11] Chlorine azide (ClN3) and bromine azide (BrN3) are extremely sensitive and explosive.[12][13]
Two series of nitrogen oxohalides are known: the nitrosyl halides (XNO) and the nitryl halides (XNO2). The first are very reactive gases that can be made by directly halogenating nitrous oxide. Nitrosyl fluoride (NOF) is colourless and a vigorous fluorinating agent. Nitrosyl fluoride can continue reacts with fluorine to form nitrogen oxide trifluoride,[14] which is also a strong fluorinating agent. Nitrosyl chloride (NOCl) behaves in much the same way and has often been used as an ionising solvent. Nitrosyl bromide (NOBr) is red. The reactions of the nitryl halides are mostly similar: nitryl fluoride (FNO2) and nitryl chloride (ClNO2) are likewise reactive gases and vigorous halogenating agents.[9]
Oxides

2 converts to colourless dinitrogen tetroxide (N
2O
4) at low temperatures, and reverts to NO
2 at higher temperatures.
Nitrogen forms nine molecular oxides, some of which were the first gases to be identified: N2O (nitrous oxide), NO (nitric oxide), N2O3 (dinitrogen trioxide), NO2 (nitrogen dioxide), N2O4 (dinitrogen tetroxide), N2O5 (dinitrogen pentoxide), N4O (nitrosylazide),[15] and N(NO2)3 (trinitramide).[16] All are thermally unstable towards decomposition to their elements. One other possible oxide that has not yet been synthesised is oxatetrazole (N4O), an aromatic ring.[15]
Nitrous oxide (N2O), better known as laughing gas, is made by thermal decomposition of molten ammonium nitrate at 250 °C. This is a redox reaction and thus nitric oxide and nitrogen are also produced as byproducts. It is mostly used as a propellant and aerating agent for sprayed canned whipped cream, and was formerly commonly used as an anaesthetic. Despite appearances, it cannot be considered to be the anhydride of hyponitrous acid (H2N2O2) because that acid is not produced by the dissolution of nitrous oxide in water. It is rather unreactive (not reacting with the halogens, the alkali metals, or ozone at room temperature, although reactivity increases upon heating) and has the unsymmetrical structure N–N–O (N≡N+O−↔−N=N+=O): above 600 °C it dissociates by breaking the weaker N–O bond.[15] Nitric oxide (NO) is the simplest stable molecule with an odd number of electrons. In mammals, including humans, it is an important cellular signaling molecule involved in many physiological and pathological processes.[17] It is formed by catalytic oxidation of ammonia. It is a colourless paramagnetic gas that, being thermodynamically unstable, decomposes to nitrogen and oxygen gas at 1100–1200 °C. Its bonding is similar to that in nitrogen, but one extra electron is added to a π* antibonding orbital and thus the bond order has been reduced to approximately 2.5; hence dimerisation to O=N–N=O is unfavourable except below the boiling point (where the cis isomer is more stable) because it does not actually increase the total bond order and because the unpaired electron is delocalised across the NO molecule, granting it stability. There is also evidence for the asymmetric red dimer O=N–O=N when nitric oxide is condensed with polar molecules. It reacts with oxygen to give brown nitrogen dioxide and with halogens to give nitrosyl halides. It also reacts with transition metal compounds to give nitrosyl complexes, most of which are deeply coloured.[15]
Blue dinitrogen trioxide (N2O3) is only available as a solid because it rapidly dissociates above its melting point to give nitric oxide, nitrogen dioxide (NO2), and dinitrogen tetroxide (N2O4). The latter two compounds are somewhat difficult to study individually because of the equilibrium between them, although sometimes dinitrogen tetroxide can react by heterolytic fission to nitrosonium and nitrate in a medium with high dielectric constant. Nitrogen dioxide is an acrid, corrosive brown gas. Both compounds may be easily prepared by decomposing a dry metal nitrate. Both react with water to form nitric acid. Dinitrogen tetroxide is very useful for the preparation of anhydrous metal nitrates and nitrato complexes, and it became the storable oxidiser of choice for many rockets in both the United States and USSR by the late 1950s. This is because it is a hypergolic propellant in combination with a hydrazine-based rocket fuel and can be easily stored since it is liquid at room temperature.[15]
The thermally unstable and very reactive dinitrogen pentoxide (N2O5) is the anhydride of nitric acid, and can be made from it by dehydration with phosphorus pentoxide. It is of interest for the preparation of explosives.[18] It is a deliquescent, colourless crystalline solid that is sensitive to light. In the solid state it is ionic with structure [NO2]+[NO3]−; as a gas and in solution it is molecular O2N–O–NO2. Hydration to nitric acid comes readily, as does analogous reaction with hydrogen peroxide giving peroxonitric acid (HOONO2). It is a violent oxidising agent. Gaseous dinitrogen pentoxide decomposes as follows:[15]
- N2O5 ⇌ NO2 + NO3 → NO2 + O2 + NO
- N2O5 + NO ⇌ 3 NO2
Oxoacids, oxoanions, and oxoacid salts
Many nitrogen oxoacids are known, though most of them are unstable as pure compounds and are known only as aqueous solution or as salts. Hyponitrous acid (H2N2O2) is a weak diprotic acid with the structure HON=NOH (pKa1 6.9, pKa2 11.6). Acidic solutions are quite stable but above pH 4 base-catalysed decomposition occurs via [HONNO]− to nitrous oxide and the hydroxide anion. Hyponitrites (involving the N
2O2−
2 anion) are stable to reducing agents and more commonly act as reducing agents themselves. They are an intermediate step in the oxidation of ammonia to nitrite, which occurs in the nitrogen cycle. Hyponitrite can act as a bridging or chelating bidentate ligand.[19]
Nitrous acid (HNO2) is not known as a pure compound, but is a common component in gaseous equilibria and is an important aqueous reagent: its aqueous solutions may be made from acidifying cool aqueous nitrite (NO−
2, bent) solutions, although already at room temperature disproportionation to nitrate and nitric oxide is significant. It is a weak acid with pKa 3.35 at 18 °C. They may be titrimetrically analysed by their oxidation to nitrate by permanganate. They are readily reduced to nitrous oxide and nitric oxide by sulfur dioxide, to hyponitrous acid with tin(II), and to ammonia with hydrogen sulfide. Salts of hydrazinium N
2H+
5 react with nitrous acid to produce azides which further react to give nitrous oxide and nitrogen. Sodium nitrite is mildly toxic in concentrations above 100 mg/kg, but small amounts are often used to cure meat and as a preservative to avoid bacterial spoilage. It is also used to synthesise hydroxylamine and to diazotise primary aromatic amines as follows:[19]
- ArNH2 + HNO2 → [ArNN]Cl + 2 H2O
Nitrite is also a common ligand that can coordinate in five ways. The most common are nitro (bonded from the nitrogen) and nitrito (bonded from an oxygen). Nitro-nitrito isomerism is common, where the nitrito form is usually less stable.[19]

Nitric acid (HNO3) is by far the most important and the most stable of the nitrogen oxoacids. It is one of the three most used acids (the other two being sulfuric acid and hydrochloric acid) and was first discovered by the alchemists in the 13th century. It is made by catalytic oxidation of ammonia to nitric oxide, which is oxidised to nitrogen dioxide, and then dissolved in water to give concentrated nitric acid. In the United States of America, over seven million tonnes of nitric acid are produced every year, most of which is used for nitrate production for fertilisers and explosives, among other uses. Anhydrous nitric acid may be made by distilling concentrated nitric acid with phosphorus pentoxide at low pressure in glass apparatus in the dark. It can only be made in the solid state, because upon melting it spontaneously decomposes to nitrogen dioxide, and liquid nitric acid undergoes self-ionisation to a larger extent than any other covalent liquid as follows:[19]
- 2 HNO3 ⇌ H
2NO+
3 + NO−
3 ⇌ H2O + [NO2]+ + [NO3]−
Two hydrates, HNO3·H2O and HNO3·3H2O, are known that can be crystallised. It is a strong acid and concentrated solutions are strong oxidising agents, though gold, platinum, rhodium, and iridium are immune to attack. A 3:1 mixture of concentrated hydrochloric acid and nitric acid, called aqua regia, is still stronger and successfully dissolves gold and platinum, because free chlorine and nitrosyl chloride are formed and chloride anions can form strong complexes. In concentrated sulfuric acid, nitric acid is protonated to form nitronium, which can act as an electrophile for aromatic nitration:[19]
- HNO3 + 2 H2SO4 ⇌ NO+
2 + H3O+ + 2 HSO−
4
The thermal stabilities of nitrates (involving the trigonal planar NO−
3 anion) depends on the basicity of the metal, and so do the products of decomposition (thermolysis), which can vary between the nitrite (for example, sodium), the oxide (potassium and lead), or even the metal itself (silver) depending on their relative stabilities. Nitrate is also a common ligand with many modes of coordination.[19]
Finally, although orthonitric acid (H3NO4), which would be analogous to orthophosphoric acid, does not exist, the tetrahedral orthonitrate anion NO3−
4 is known in its sodium and potassium salts:[19]
![{\displaystyle {\ce {NaNO3{}+Na2O->[{\ce {Ag~crucible}}][{\ce {300^{\circ }C~for~7days}}]Na3NO4}}}](../I/ec729bc88f520e08fdce8a013dec8ae601d28509.svg)
These white crystalline salts are very sensitive to water vapour and carbon dioxide in the air:[19]
- Na3NO4 + H2O + CO2 → NaNO3 + NaOH + NaHCO3
Despite its limited chemistry, the orthonitrate anion is interesting from a structural point of view due to its regular tetrahedral shape and the short N–O bond lengths, implying significant polar character to the bonding.[19]
Organic nitrogen compounds
Nitrogen is one of the most important elements in organic chemistry. Many organic functional groups involve a carbon–nitrogen bond, such as amides (RCONR2), amines (R3N), imines (RC(=NR)R), imides (RCO)2NR, azides (RN3), azo compounds (RN2R), cyanates and isocyanates (ROCN or RCNO), nitrates (RONO2), nitriles and isonitriles (RCN or RNC), nitrites (RONO), nitro compounds (RNO2), nitroso compounds (RNO), oximes (RCR=NOH), and pyridine derivatives. C–N bonds are strongly polarised towards nitrogen. In these compounds, nitrogen is usually trivalent (though it can be tetravalent in quaternary ammonium salts, R4N+), with a lone pair that can confer basicity on the compound by being coordinated to a proton. This may be offset by other factors: for example, amides are not basic because the lone pair is delocalised into a double bond (though they may act as acids at very low pH, being protonated at the oxygen), and pyrrole is not acidic because the lone pair is delocalised as part of an aromatic ring.[20] The amount of nitrogen in a chemical substance can be determined by the Kjeldahl method.[21] In particular, nitrogen is an essential component of nucleic acids, amino acids and thus proteins, and the energy-carrying molecule adenosine triphosphate and is thus vital to all life on Earth.[20]
See also
References
- Fryzuk, M. D. & Johnson, S. A. (2000). "The continuing story of dinitrogen activation". Coordination Chemistry Reviews. 200–202: 379. doi:10.1016/S0010-8545(00)00264-2.
- Schrock, R. R. (2005). "Catalytic Reduction of Dinitrogen to Ammonia at a Single Molybdenum Center". Acc. Chem. Res. 38 (12): 955–62. doi:10.1021/ar0501121. PMC 2551323. PMID 16359167.
- Greenwood, Norman N.; Earnshaw, Alan (1997). Chemistry of the Elements (2nd ed.). Butterworth-Heinemann. ISBN 978-0-08-037941-8.
- Greenwood and Earnshaw, pp. 417–20
- Greenwood and Earnshaw, pp. 434–38
- Greenwood and Earnshaw, pp. 420–26
- Greenwood and Earnshaw, pp. 426–33
- Vieira, R.; C. Pham-Huu; N. Keller; M. J. Ledoux (2002). "New carbon nanofiber/graphite felt composite for use as a catalyst support for hydrazine catalytic decomposition". Chemical Communications (9): 954–55. doi:10.1039/b202032g. PMID 12123065.
- Greenwood and Earnshaw, pp. 438–42
- Bowden, F. P. (1958). "Initiation of Explosion by Neutrons, α-Particles, and Fission Products". Proceedings of the Royal Society of London A. 246 (1245): 216–19. Bibcode:1958RSPSA.246..216B. doi:10.1098/rspa.1958.0123. S2CID 137728239.
- Ford, L. A.; Grundmeier, E. W. (1993). Chemical Magic. Dover. p. 76. ISBN 978-0-486-67628-9.
- Frierson, W. J.; Kronrad, J.; Browne, A. W. (1943). "Chlorine Azide, ClN3. I". Journal of the American Chemical Society. 65 (9): 1696–1698. doi:10.1021/ja01249a012.
- Lyhs, Benjamin; Bläser, Dieter; Wölper, Christoph; Schulz, Stephan; Jansen, Georg (20 February 2012). "Solid-State Structure of Bromine Azide" (PDF). Angewandte Chemie International Edition. 51 (8): 1970–1974. doi:10.1002/anie.201108092. PMID 22250068. Archived (PDF) from the original on 25 August 2021. Retrieved 25 August 2021.
- Fox, W.B.; MacKenzie, J.S.; Vitek, R. (February 1970). "The chemistry of trifluoramine oxide. V. Synthesis of F3 no by photochemical fluorination of nitrosyl fluoride". Inorganic and Nuclear Chemistry Letters. 6 (2): 177–179. doi:10.1016/0020-1650(70)80336-1.
- Greenwood and Earnshaw, pp. 443–58
- Rahm, Martin; Dvinskikh, Sergey V.; Furó, István; Brinck, Tore (23 December 2010). "Experimental Detection of Trinitramide, N(NO2)3". Angewandte Chemie International Edition. 50 (5): 1145–48. doi:10.1002/anie.201007047. PMID 21268214. S2CID 32952729.
- Hou, Y. C.; Janczuk, A.; Wang, P. G. (1999). "Current trends in the development of nitric oxide donors". Current Pharmaceutical Design. 5 (6): 417–41. doi:10.2174/138161280506230110111042. PMID 10390607.
- Talawar, M. B.; et al. (2005). "Establishment of Process Technology for the Manufacture of Dinitrogen Pentoxide and its Utility for the Synthesis of Most Powerful Explosive of Today – CL-20". Journal of Hazardous Materials. 124 (1–3): 153–64. doi:10.1016/j.jhazmat.2005.04.021. PMID 15979786.
- Greenwood and Earnshaw, pp. 459–72
- March, Jerry (1985), Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 3rd edition, New York: Wiley, ISBN 9780471854722, OCLC 642506595
- Rédei, George P (2008). "Kjeldahl Method". Encyclopedia of Genetics, Genomics, Proteomics and Informatics. p. 1063. doi:10.1007/978-1-4020-6754-9_9066. ISBN 978-1-4020-6753-2.