Omigapil
Omigapil (TCH346 or CGP3466) is a drug that was developed by Novartis and tested in clinical trials for its ability to help treat Parkinson's disease (PD)[1] and amyotrophic lateral sclerosis (ALS).[2] The development for PD and ALS have been terminated due to lack of benefit, but Santhera Pharmaceuticals bought the compound for development for the treatment of congenital muscular dystrophy (CMD).[3][4][5][6]
 | |
| Clinical data | |
|---|---|
| Other names | TCH346, CGP3466B |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| CompTox Dashboard (EPA) | |
| Chemical and physical data | |
| Formula | C19H17NO |
| Molar mass | 275.351 g·mol−1 |
| 3D model (JSmol) | |
| |
| |
Omigapil was first synthesized at Ciba-Geigy, Basel, Switzerland. Santhera Pharmaceuticals has since taken over production of omigapil and preclinical trials for CMD. In May 2008, omigapil was granted orphan designation to commence clinical trials for.[7] Pharmacokinetic trials are scheduled to commence enrollment in the second half of 2012 to determine the appropriate pharmacokinetic profile of the drug for children with laminin-α2-deficient congenital muscular dystrophy (MDC1A) and collagen VI related myopathy. Santhera Pharmaceuticals will use the phase 1 clinical trial to determine if the drug is safe and acts with the same pharmacokinetic profile in children as it does in adults. The impending clinical trial will take place in the United States at the National Institute of Neurological Disorders and Stroke/National Institute of Health(NNDCS/NINDS) (Bethesda, Maryland) and in the United Kingdom at Great Ormond Street Hospital (UCL).[8]
Mechanism of action
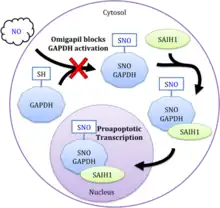
Omigapil inhibits programmed cell death (apoptosis) through the enzymes glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and SIAH1. The glycolytic housekeeping enzyme GAPDH is mediated by neuronal nitric oxide synthase.[9] Once activated by nitric oxide, GAPDH binds to the ubiquitin ligase SIAH1, and is transported to the nucleus where it activates the acetyltransferases p300/CBP to enhance acetylation and subsequent transcription. GAPDH's targets proapoptotic genes such as p53, p53 upregulated modulator of apoptosis (PUMA), and p21 as well as other related targets.[10] Chemogenetic studies indicate that omigapil inhibits this proapoptotic signaling cascade by preventing GAPDH activation through S-nitrosylation, which in turn prevents the binding of SIAH1 and translocation to the nucleus (see figure).[11] Multiple binding cites on GAPDH have been suggested.[12]
Omigapil was originally developed as a structurally similar molecule to selegiline (L-deprenyl), a monoamine oxidase inhibitor (MAO) blocking the enzyme MAO type B, yet omigapil inhibit neither type of MAO.[13] Selegiline has proven problematic as a treatment for Parkinson's disease because it is metabolized to (meth)amphetamine, which gives rise to adverse effects. Due to omigapil's tricyclic nature, the drug cannot be metabolized to amphetamine derivatives.[14] Omigapil acts as a neuroprotective agent in cellular and rodent models of Parkinson's disease like selegiline, but its neuroprotective action is 100 times more potent than selegiline in both in vivo and in vitro studies.[15]
Pharmacokinetics
Omigapil can pass through the blood brain barrier and has oral bioavailability as omigapil mono-maleate salt.[16] Studies have demonstrated a bell-shaped dose-response curve for both rodent and primate models. The rhesus monkey dose was optimized between 0.014 and 0.14 mg/kg subcutaneous.[17] In human trials for Parkinson's disease, doses of 0.5, 2.5 and 10 mg daily were considered, which resulted in the selection of a dose range of 0.3 to 3 mg daily for a 70 kg individual.[14] Unfortunately a biomarker has not been established for omigapil, which means that clinical trials rely on blood plasma levels to measure drug distribution rather than a validated biomarker to specifically measure brain exposure.[5][14]
Efficacy in animal models
The compound displayed cell-rescuing effects in various models of apoptotic neuronal death, as well as in rodent and non-rodent animal models of neurodegeneration. Omigapil rescues in vitro PC12 cells from rotenone toxicity, β-amyloid toxicity, nutrition withdrawal, and lactacystin.[14] Additionally, omigapil can prevent NMDA and kainate receptor excitotoxicity in rat cortical neurons as well as toxicity from cytosine arabinoside (ara C) in cerebellar granule cells. Omigapil also rescues rat oligodendrocytes from AMPA receptor excitotoxicity and rat embryonic mesencephalic (midbrain) dopaminergic cells from toxicity by MPP+/MPTP.[18] In human neuroblastoma (PAJU) cells, omigapil can also prevent toxicity from rotenone and GAPDH overexpression. Omigapil has an active concentration range from about 10−12 M to 10−5 M, with a maximum at about 10−9 M. Omigapil prevents neurodegeneration in facial motor neuron axotomy animal models as well as mouse models of progressive motor neuronopathy, MPTP-induced nigrostriatal degeneration, and oxidopamine-induced neuronal injury.[15] Omigapil also prevents the death of nigrostriatal dopaminergic neurons in monkeys treated with MPTP to mimic Parkinson's disease symptoms.[17] While omigapil was able to prevent programmed cell death for high-risk cells and prevent deterioration of concomitant motor deficits associated with Parkinson's symptoms, omigapil was unable to reverse pre-existing Parkinson's symptoms in MPTP monkeys.[17]
Clinical trials
Parkinson's disease and ALS
Based on the preclinical results mentioned above, clinical trials were run for both Parkinson's disease and amyotrophic lateral sclerosis, but omigapil proved to be inefficacious for both diseases.[5] It is unclear whether the discrepancy in results between laboratory studies and clinical studies is from improper pathogenesis modeling of the disease in animal models, insufficient doses of the study drug, insensitive clinical endpoints, or abnormal sampling in the patient population. However, the drug was determined to be safe for human use with no notable serious side effects.[5]
Research
Congenital muscular dystrophy
Omigapil can ameliorate congenital muscular dystrophy (CMD) symptoms.[19] This rare yet fatal infant disease has symptoms ranging from severe neonatal hypotonia ("floppy infant syndrome") to peripheral neuropathy, inability to stand or walk, respiratory distress, and eventually premature death in early life. The majority of CMD cases result from a genetic mutation in laminin-α2, a subunit of the laminin-211 protein, which serves as an essential mechanical link between basement membrane and muscle fiber in skeletal and heart muscle.[20] The result is muscle degeneration and demyelination of peripheral nerves.[21]
The mouse model of laminin-α2-deficient congenital muscular dystrophy (MDC1A) was found to positively respond to omigapil with inhibition of apoptosis in muscle, reduction of body weight loss and skeletal deformation, increased locomotive activity, and protection from early mortality.[22] Furthermore, omigapil was found to be even more effective in improving muscle function and strength when coupled with overexpression of the extracellular matrix molecule mini-agrin in MDC1A mice.[23] Omigapil coupled with mini-agrin overexpression works as a dual treatment that enhances mechanical load bearing ability and improves regeneration of muscle in MDC1A mice. Given that the technology for mini-agrin administration to skeletal muscle in human subjects is not yet available, omigapil is ready for human clinical trials to help mediate CMD. Omigapil has undergone extensive clinical trial scrutiny for Parkinson's disease and ALS, which indicates that the drug is safe to begin clinical trials for congenital muscular dystrophy.[5][24]
References
- Olanow, C. W.; Schapira, A. H.; Lewitt, P. A.; Kieburtz, K.; Sauer, D.; Olivieri, G.; Pohlmann, H.; Hubble, J. (December 2006). "TCH346 as a neuroprotective drug in Parkinson's disease: a double-blind, randomised, controlled trial". Lancet Neurol. 5 (12): 1013–1020. doi:10.1016/S1474-4422(06)70602-0. PMID 17110281. S2CID 1562331.
- Clinical trial number NCT00036413 for "A 12-Week, Multicenter, Safety and Dose-Ranging Study of 3 Oral Doses of TCH346 in Patients With Amyotrophic Lateral Sclerosis" at ClinicalTrials.gov
- "Santhera to Test Compound in CMD". Archived from the original on 2011-10-25. Retrieved 2011-09-18.
- Meinen S, Lin S, Thurnherr R, Erb M, Meier T, Rüegg MA (2011). "Apoptosis inhibitors and mini-agrin have additive benefits in congenital muscular dystrophy mice". EMBO Molecular Medicine. 3 (8): 465–479. doi:10.1002/emmm.201100151. PMC 3377088. PMID 21674808.
- Olanow CW, Schapira AH, LeWitt PA, Kieburtz K, Sauer D, Olivieri G, Pohlmann H, Hubble J (December 2006). "TCH346 as a neuroprotective drug in Parkinson's disease: a double-blind, randomised, controlled trial". Lancet Neurol. 5 (12): 1013–20. doi:10.1016/S1474-4422(06)70602-0. PMID 17110281. S2CID 1562331.
- "Santhera Pharmaceuticals: Development of SNT-317 (INN: omigapil) in CMD and other neuromuscular diseases" (PDF). Archived from the original (PDF) on 2016-03-04. Retrieved 2012-05-07.
- Committee for Orphan Medicinal Products. "Public summary of opinion on orphan designation: Omigapil maleate for the congenital muscular dystrophy with merosin (laminin alpha 2) deficiency" (PDF). EMA/COMP/204694/2008 Rev.1.
- Muscular Dystrophy Campaign. "New collaboration to support omigapil clinical trial for congenital muscular dystrophy". Retrieved 13 May 2012.
- Hara MR, Agrawal N, Kim SF, Cascio MB, Fujimuro M, Ozeki Y, Takahashi M, Cheah JH, Tankou SK, Hester LD, Ferris CD, Hayward SD, Snyder SH, Sawa A (July 2005). "S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding". Nat. Cell Biol. 7 (7): 665–74. doi:10.1038/ncb1268. PMID 15951807. S2CID 1922911.
- Sen N, Hara MR, Kornberg MD, Cascio MB, Bae BI, Shahani N, Thomas B, Dawson TM, Dawson VL, Snyder SH, Sawa A (July 2008). "Nitric oxide-induced nuclear GAPDH activates p300/CBP and mediates apoptosis". Nat. Cell Biol. 10 (7): 866–73. doi:10.1038/ncb1747. PMC 2689382. PMID 18552833.
- Hara MR, Thomas B, Cascio MB, Bae BI, Hester LD, Dawson VL, Dawson TM, Sawa A, Snyder SH (March 2006). "Neuroprotection by pharmacologic blockade of the GAPDH death cascade". Proc. Natl. Acad. Sci. U.S.A. 103 (10): 3887–9. Bibcode:2006PNAS..103.3887H. doi:10.1073/pnas.0511321103. PMC 1450161. PMID 16505364.
- Jenkins JL, Tanner JJ (March 2006). "High-resolution structure of human D-glyceraldehyde-3-phosphate dehydrogenase". Acta Crystallogr. D. 62 (Pt 3): 290–301. doi:10.1107/S0907444905042289. PMID 16510976.
- Kragten E, Lalande I, Zimmermann K, Roggo S, Schindler P, Muller D, van Oostrum J, Waldmeier P, Furst P (March 1998). "Glyceraldehyde-3-phosphate dehydrogenase, the putative target of the antiapoptotic compounds CGP 3466 and R-(−)-deprenyl". J. Biol. Chem. 273 (10): 5821–8. doi:10.1074/jbc.273.10.5821. PMID 9488718.
- Waldmeier P, Bozyczko-Coyne D, Williams M, Vaught JL (November 2006). "Recent clinical failures in Parkinson's disease with apoptosis inhibitors underline the need for a paradigm shift in drug discovery for neurodegenerative diseases". Biochem. Pharmacol. 72 (10): 1197–206. doi:10.1016/j.bcp.2006.06.031. PMID 16901468.
- Waldmeier PC, Boulton AA, Cools AR, Kato AC, Tatton WG (2000). "Neurorescuing effects of the GAPDH ligand CGP 3466B". J. Neural Transm. Suppl. (60): 197–214. doi:10.1007/978-3-7091-6301-6_13. ISBN 978-3-211-83537-1. PMID 11205140.
- Sagot, Y; Toni, N; Perrelet, D; Lurot, S; King, B; Rixner, H; Mattenberger, L; Waldmeier, P C; Kato, A C (1 October 2000). "An orally active anti-apoptotic molecule (CGP 3466B) preserves mitochondria and enhances survival in an animal model of motoneuron disease". British Journal of Pharmacology. 131 (4): 721–728. doi:10.1038/sj.bjp.0703633. PMC 1572390. PMID 11030721.
- Andringa G, Eshuis S, Perentes E, Maguire RP, Roth D, Ibrahim M, Leenders KL, Cools AR (November 2003). "TCH346 prevents motor symptoms and loss of striatal FDOPA uptake in bilaterally MPTP-treated primates". Neurobiol. Dis. 14 (2): 205–17. doi:10.1016/S0969-9961(03)00125-6. PMID 14572443. S2CID 25987945.
- Andringa G, Cools AR (2000). "The neuroprotective effects of CGP 3466B in the best in vivo model of Parkinson's disease, the bilaterally MPTP-treated rhesus monkey". J. Neural Transm. Suppl. (60): 215–25. doi:10.1007/978-3-7091-6301-6_14. ISBN 978-3-211-83537-1. PMID 11205142.
- Erb, M.; Meinen, S.; Barzaghi, P.; Sumanovski, L. T.; Courdier-Fruh, I.; Ruegg, M. A.; Meier, T. (16 September 2009). "Omigapil Ameliorates the Pathology of Muscle Dystrophy Caused by Laminin- 2 Deficiency". Journal of Pharmacology and Experimental Therapeutics. 331 (3): 787–795. doi:10.1124/jpet.109.160754. PMID 19759319. S2CID 26038408.
- Colognato H, Yurchenco PD (June 2000). "Form and function: the laminin family of heterotrimers". Dev. Dyn. 218 (2): 213–34. doi:10.1002/(SICI)1097-0177(200006)218:2<213::AID-DVDY1>3.0.CO;2-R. PMID 10842354. S2CID 33567462.
- Miyagoe-Suzuki Y, Nakagawa M, Takeda S (2000). "Merosin and congenital muscular dystrophy". Microsc. Res. Tech. 48 (3–4): 181–91. doi:10.1002/(SICI)1097-0029(20000201/15)48:3/4<181::AID-JEMT6>3.0.CO;2-Q. PMID 10679965. S2CID 23239499.
- Erb M, Meinen S, Barzaghi P, Sumanovski LT, Courdier-Früh I, Rüegg MA, Meier T (December 2009). "Omigapil ameliorates the pathology of muscle dystrophy caused by laminin-alpha2 deficiency". J. Pharmacol. Exp. Ther. 331 (3): 787–95. doi:10.1124/jpet.109.160754. PMID 19759319. S2CID 26038408.
- Meinen S, Lin S, Thurnherr R, Erb M, Meier T, Rüegg MA (August 2011). "Apoptosis inhibitors and mini-agrin have additive benefits in congenital muscular dystrophy mice". EMBO Molecular Medicine. 3 (8): 465–79. doi:10.1002/emmm.201100151. PMC 3377088. PMID 21674808.
- Miller R, Bradley W, Cudkowicz M, Hubble J, Meininger V, Mitsumoto H, Moore D, Pohlmann H, Sauer D, Silani V, Strong M, Swash M, Vernotica E (August 2007). "Phase II/III randomized trial of TCH346 in patients with ALS". Neurology. 69 (8): 776–84. doi:10.1212/01.wnl.0000269676.07319.09. PMID 17709710. S2CID 29883238.
- Harraz, Maged M.; Tyagi, Richa; Cortés, Pedro; Snyder, Solomon H. (2016-10-23). "Antidepressant action of ketamine via mTOR is mediated by inhibition of nitrergic Rheb degradation". Molecular Psychiatry. 21 (3): 313–319. doi:10.1038/mp.2015.211. ISSN 1359-4184. PMC 4830355. PMID 26782056.
Further reading
- Andringa G, Eshuis S, Perentes E, Maguire RP, Roth D, Ibrahim M, Leenders KL, Cools AR (2003). "TCH346 prevents motor symptoms and loss of striatal FDOPA uptake in bilaterally MPTP-treated primates". Neurobiology of Disease. 14 (2): 205–217. doi:10.1016/S0969-9961(03)00125-6. PMID 14572443. S2CID 25987945.