PDE6B
Rod cGMP-specific 3',5'-cyclic phosphodiesterase subunit beta is the beta subunit of the protein complex PDE6 that is encoded by the PDE6B gene.[5][6] PDE6 is crucial in transmission and amplification of visual signal. The existence of this beta subunit is essential for normal PDE6 functioning. Mutations in this subunit are responsible for retinal degeneration such as retinitis pigmentosa[7][8] or congenital stationary night blindness.[9]



Structure
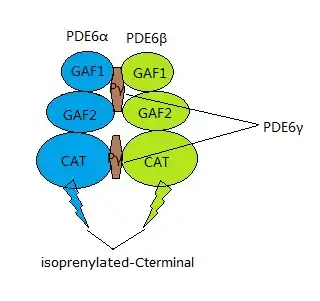
PDE6 is a protein complex located on the photoreceptor's outer segment, and plays an important role in the phototransduction cascade.[10] There are two types of photoreceptors: cones and rods. The rod and cone PDE6 complexes have different structures. PDE6β together with PDE6α and two identical inhibitory subunits, PDE6γ, form the rod PDE6 holoenzyme[11] while the cone PDE6 complex only consists of two identical PDE6α' catalytic subunits.[12] PDE6β, one of the catalytic units in rod PDE6, is composed of three domains: two N-terminal GAF domains and one C-terminal catalytic domain.[13] The non-catalytic GAF domains are responsible for cGMP binding. The C-terminal interacts with cell membrane by isoprenylation and S-carboxylmethylation.[12]
Function
Absorption of photons by rhodopsin triggers a signal transduction cascade in rod photoreceptors. This phototransduction cascade leads to hydrolysis of cGMP by cGMP-phosphodiesterase (PDE) that closes cGMP-gated channels and hyperpolarizes the cell.[14] PDE6β is necessary for the formation of a functional phosphodiesterase holoenzyme.[12]
Function of PDE6
PDE6 is a highly concentrated protein in retinal photoreceptors. With the presence of the GAF domain, PDE6 can actively bind to the cGMP. The inactive PDE6 in the dark allows cGMP to bind to cGMP gated ion channels. The channel remains open as long as cGMP is binding to it, which allows constant electron flow in to the photoreceptor cell through the plasma membrane. Light causes the visual pigment, rhodopsin, to activate. This process leads to the release of subunit PDE6γ from PDE6αβ, activating PDE6 which leads to the hydrolysis of cGMP. Without the cGMP binding, the ion channel closes, leading to the hyperpolarization.[12] After hyperpolarization the presnaptic transmitter is reduced. Next, the enzyme guanylate cyclase restores cGMP, which reopens the membrane channels. This process is called light adaptation.
Function of PDE6B
PDE6β is the only protein that undergoes the two types of post-translational modification, prenylation and carboxymethylation.[15] The geranylgeranyl group of PDE6B is the result of these modifications, which are responsible for the rod PDE6's interaction with membrane.
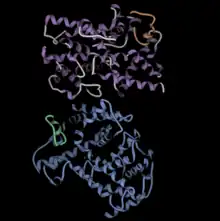
The figure at left shows the PDE6 aalpha/beta dimer in blue and purple, with the gamma ubunits in green and orange.
Animal studies
rd1 mouse
Mutation of the PDE6b gene leads to the dysfunction of PDE, which results in failure of hydrolysis of cGMP. The rd1 mouse is a well-characterized animal model of retinitis pigmentosa caused by the mutation of Pde6b gene.[16] The phenotype was first discovered in rodless mice in the 1920s by Keeler.[17] An insertion of Murine leukemia provirus is present near the first exon combined with a point mutation, which introduces a stop codon in exon 7. In addition to the rd1 mouse, a missense mutation (R560C) in exon 13 of the Pde6b gene is the character of another animal model of recessive retinal degeneration.
In rd1 animals, the retinal rod photoreceptor cells begin degenerating at about postnatal day 10, and by 3 weeks no rod photoreceptors remain. Degeneration is preceded by accumulation of cGMP in the retina and is correlated with deficient activity of the rod photoreceptor cGMP-phosphodiesterase.[16][18] Cone photoreceptors undergo a slower degeneration over the course of a year, which causes the mutants to completely go blind.[19] The possibility of altering the course of retinal degeneration through subretinal injection of recombinant replication defective adenovirus that contained the murine cDNA for wildtype PDE6β was tested in rd1 mice.[20] Subretinal injection of rd1 mice was carried out 4 days after birth, before the onset of rod photoreceptor degeneration. Following therapy, Pde6β transcripts and enzyme activity were detected, and histologic studies revealed that photoreceptor cell death was significantly retarded.[6]
The albino FVB mouse laboratory strain become blind by weaning age due to a mutant allele of the PDE6b gene. There are pigmented derivative strains of FVB that lack this trait.
rcd1 dog
Similar to rd1 in mice, Rod-cone dysplasia type 1 (rcd1-PRA) is a form of progressive retinal atrophy (PRA), with early onset of the disease. The Irish Setter is a characterized animal model of rcd1. The mutation is caused by a nonsense mutation in pde6b gene. Photoreceptors start degeneration at postnatal day 13 until a year after the dog is totally blind.[21]
References
- GRCh38: Ensembl release 89: ENSG00000133256 - Ensembl, May 2017
- GRCm38: Ensembl release 89: ENSMUSG00000029491 - Ensembl, May 2017
- "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- Bateman JB, Klisak I, Kojis T, Mohandas T, Sparkes RS, Li TS, Applebury ML, Bowes C, Farber DB (Mar 1992). "Assignment of the beta-subunit of rod photoreceptor cGMP phosphodiesterase gene PDEB (homolog of the mouse rd gene) to human chromosome 4p16". Genomics. 12 (3): 601–3. doi:10.1016/0888-7543(92)90454-Z. PMID 1313787.
- "Entrez Gene: PDE6B phosphodiesterase 6B, cGMP-specific, rod, beta (congenital stationary night blindness 3, autosomal dominant)".
- Wang Q, Chen Q, Zhao K, Wang L, Wang L, Traboulsi EI (2001). "Update on the molecular genetics of retinitis pigmentosa". Ophthalmic Genetics. 22 (3): 133–54. doi:10.1076/opge.22.3.133.2224. PMID 11559856. S2CID 24004113.
- Danciger, M; Blaney, J; Gao, YQ; Zhao, DY; Heckenlively, JR; Jacobson, SG; Farber, DB (1 November 1995). "Mutations in the PDE6B gene in autosomal recessive retinitis pigmentosa". Genomics. 30 (1): 1–7. doi:10.1006/geno.1995.0001. PMID 8595886.
- "RetNet: Genes and Mapped Loci Causing Retinal Diseases". RetNet. Retrieved 12 May 2015.
- Han J, Dinculescu A, Dai X, Du W, Smith WC, Pang J (20 December 2013). "Review: the history and role of naturally occurring mouse models with Pde6b mutations". Molecular Vision. 19: 2579–89. PMC 3869645. PMID 24367157.
- Gulati S, Palczewski K, Engel A, Stahlberg H, Kovacik L (Feb 2019). "Cryo-EM structure of phosphodiesterase 6 reveals insights into the allosteric regulation of type I phosphodiesterases". Science Advances. 27 (5): eaav4322. doi:10.1126/sciadv.aav4322. PMC 6392808. PMID 30820458.
- Cote RH (Jun 2004). "Characteristics of photoreceptor PDE (PDE6): similarities and differences to PDE5". International Journal of Impotence Research. 16 Suppl 1: S28-33. doi:10.1038/sj.ijir.3901212. PMID 15224133.
- Gulati S, Palczewski K, Engel A, Stahlberg H, Kovacik L (Feb 2019). "Cryo-EM structure of phosphodiesterase 6 reveals insights into the allosteric regulation of type I phosphodiesterases". Science Advances. 27 (5): eaav4322. doi:10.1126/sciadv.aav4322. PMC 6392808. PMID 30820458.
- Khramtsov NV, Feshchenko EA, Suslova VA, Shmukler BE, Terpugov BE, Rakitina TV, Atabekova NV, Lipkin VM (Aug 1993). "The human rod photoreceptor cGMP phosphodiesterase beta-subunit. Structural studies of its cDNA and gene". FEBS Letters. 327 (3): 275–8. doi:10.1016/0014-5793(93)81003-I. PMID 8394243. S2CID 221433455.
- Anant JS, Ong OC, Xie HY, Clarke S, O'Brien PJ, Fung BK (Jan 1992). "In vivo differential prenylation of retinal cyclic GMP phosphodiesterase catalytic subunits". The Journal of Biological Chemistry. 267 (2): 687–90. doi:10.1016/S0021-9258(18)48336-6. PMID 1309771.
- Farber DB, Lolley RN (1976). "Enzymic basis for cyclic GMP accumulation in degenerative photoreceptor cells of mouse retina". Journal of Cyclic Nucleotide Research. 2 (3): 139–48. PMID 6493.
- Keeler, CE (20 March 1928). "The Geotropic Reaction of Rodless Mice in Light and in Darkness". The Journal of General Physiology. 11 (4): 361–8. doi:10.1085/jgp.11.4.361. PMC 2140980. PMID 19872404.
- Farber DB, Lolley RN (Nov 1974). "Cyclic guanosine monophosphate: elevation in degenerating photoreceptor cells of the C3H mouse retina". Science. 186 (4162): 449–51. doi:10.1126/science.186.4162.449. PMID 4369896. S2CID 44714968.
- Chang B, Hawes NL, Hurd RE, Davisson MT, Nusinowitz S, Heckenlively JR (Feb 2002). "Retinal degeneration mutants in the mouse". Vision Research. 42 (4): 517–25. doi:10.1016/s0042-6989(01)00146-8. PMID 11853768. S2CID 17442038.
- Bennett J, Tanabe T, Sun D, Zeng Y, Kjeldbye H, Gouras P, Maguire AM (Jun 1996). "Photoreceptor cell rescue in retinal degeneration (rd) mice by in vivo gene therapy". Nature Medicine. 2 (6): 649–54. doi:10.1038/nm0696-649. PMID 8640555. S2CID 9184060.
- Petit L, Lhériteau E, Weber M, Le Meur G, Deschamps JY, Provost N, Mendes-Madeira A, Libeau L, Guihal C, Colle MA, Moullier P, Rolling F (Nov 2012). "Restoration of vision in the pde6β-deficient dog, a large animal model of rod-cone dystrophy". Molecular Therapy. 20 (11): 2019–30. doi:10.1038/mt.2012.134. PMC 3498794. PMID 22828504.
Further reading
- Lerner LE, Piri N, Farber DB (2007). Transcriptional and post-transcriptional regulation of the rod cGMP-phosphodiesterase beta-subunit gene. Recent advances and current concepts. Advances in Experimental Medicine and Biology. Vol. 572. pp. 217–29. doi:10.1007/0-387-32442-9_32. ISBN 978-0-387-28464-4. PMID 17249578.
- Altherr MR, Wasmuth JJ, Seldin MF, Nadeau JH, Baehr W, Pittler SJ (Apr 1992). "Chromosome mapping of the rod photoreceptor cGMP phosphodiesterase beta-subunit gene in mouse and human: tight linkage to the Huntington disease region (4p16.3)". Genomics. 12 (4): 750–4. doi:10.1016/0888-7543(92)90305-C. PMID 1315306.
- Collins C, Hutchinson G, Kowbel D, Riess O, Weber B, Hayden MR (Jul 1992). "The human beta-subunit of rod photoreceptor cGMP phosphodiesterase: complete retinal cDNA sequence and evidence for expression in brain". Genomics. 13 (3): 698–704. doi:10.1016/0888-7543(92)90144-H. PMID 1322354.
- Catty P, Pfister C, Bruckert F, Deterre P (Sep 1992). "The cGMP phosphodiesterase-transducin complex of retinal rods. Membrane binding and subunits interactions". The Journal of Biological Chemistry. 267 (27): 19489–93. doi:10.1016/S0021-9258(18)41802-9. PMID 1326553.
- Khramtsov NV, Feshchenko EA, Suslova VA, Terpugov BE, Rakitina TV, Atabekova NV, Shmukler BE, Lipkin VM (Dec 1992). "[Structural studies of cDNA and the gene for the beta-subunit of cGMP phosphodiesterase from human retina]". Bioorganicheskaia Khimiia. 18 (12): 1551–4. PMID 1338685.
- Weber B, Riess O, Hutchinson G, Collins C, Lin BY, Kowbel D, Andrew S, Schappert K, Hayden MR (Nov 1991). "Genomic organization and complete sequence of the human gene encoding the beta-subunit of the cGMP phosphodiesterase and its localisation to 4p 16.3". Nucleic Acids Research. 19 (22): 6263–8. doi:10.1093/nar/19.22.6263. PMC 329137. PMID 1720239.
- McLaughlin ME, Ehrhart TL, Berson EL, Dryja TP (Apr 1995). "Mutation spectrum of the gene encoding the beta subunit of rod phosphodiesterase among patients with autosomal recessive retinitis pigmentosa". Proceedings of the National Academy of Sciences of the United States of America. 92 (8): 3249–53. doi:10.1073/pnas.92.8.3249. PMC 42143. PMID 7724547.
- Gal A, Orth U, Baehr W, Schwinger E, Rosenberg T (Aug 1994). "Heterozygous missense mutation in the rod cGMP phosphodiesterase beta-subunit gene in autosomal dominant stationary night blindness". Nature Genetics. 7 (4): 551. doi:10.1038/ng0894-551a. PMID 7951329.
- Gal A, Orth U, Baehr W, Schwinger E, Rosenberg T (May 1994). "Heterozygous missense mutation in the rod cGMP phosphodiesterase beta-subunit gene in autosomal dominant stationary night blindness". Nature Genetics. 7 (1): 64–8. doi:10.1038/ng0594-64. PMID 8075643. S2CID 33020561.
- McLaughlin ME, Sandberg MA, Berson EL, Dryja TP (Jun 1993). "Recessive mutations in the gene encoding the beta-subunit of rod phosphodiesterase in patients with retinitis pigmentosa". Nature Genetics. 4 (2): 130–4. doi:10.1038/ng0693-130. PMID 8394174. S2CID 10406814.
- Valverde D, Solans T, Grinberg D, Balcells S, Vilageliu L, Bayés M, Chivelet P, Besmond C, Goossens M, González-Duarte R, Baiget M (Jan 1996). "A novel mutation in exon 17 of the beta-subunit of rod phosphodiesterase in two RP sisters of a consanguineous family". Human Genetics. 97 (1): 35–8. doi:10.1007/BF00218829. PMID 8557257. S2CID 9003586.
- Gao YQ, Danciger M, Zhao DY, Blaney J, Piriev NI, Shih J, Jacobson SG, Heckenlively JH, Farber DB (Feb 1996). "Screening of the PDE6B gene in patients with autosomal dominant retinitis pigmentosa". Experimental Eye Research. 62 (2): 149–54. doi:10.1006/exer.1996.0019. PMID 8698075.
- Suslova VA, Suslov ON, Kim EE, Lipkin VM (Apr 1996). "[Organization of the gene for the beta-subunit of human photoreceptor cyclic GMP phosphodiesterase]". Bioorganicheskaia Khimiia. 22 (4): 256–63. PMID 8768262.
- Valverde D, Baiget M, Seminago R, del Rio E, García-Sandoval B, del Rio T, Bayés M, Balcells S, Martínez A, Grinberg D, Ayuso C (1997). "Identification of a novel R552O mutation in exon 13 of the beta-subunit of rod phosphodiesterase gene in a Spanish family with autosomal recessive retinitis pigmentosa". Human Mutation. 8 (4): 393–4. doi:10.1002/humu.1380080403. PMID 8956055. S2CID 84385812.