Pemphigoid
Pemphigoid is a group of rare autoimmune blistering diseases of the skin, and mucous membranes. As its name indicates, pemphigoid is similar in general appearance to pemphigus,[1] but, unlike pemphigus, pemphigoid does not feature acantholysis, a loss of connections between skin cells.[2]
| Pemphigoid | |
|---|---|
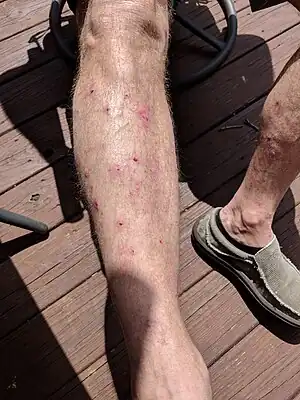 | |
| Vesicles and bullae shown on the lower leg, some ruptured leaving a crusted area in bullous pemphigoid | |
| Specialty | Dermatology |
Pemphigoid is more common than pemphigus, and is slightly more common in women than in men. It is also more common in people aged over 70 years than it is in younger people.[3]
Classification
IgG
The forms of pemphigoid are considered to be connective tissue autoimmune skin diseases. There are several types:
- Gestational pemphigoid (PG) (formerly called Herpes gestationis)
- Bullous pemphigoid (BP) Rarely affects the mouth
- Mucous membrane pemphigoid (MMP) or (cicatricial pemphigoid), (No skin involvement)
Bullous and mucous membrane pemphigoid usually affect persons who are over age 60.[4][5] Gestational pemphigoid occurs during pregnancy,[6] typically in the second or third trimester, or immediately following pregnancy.
Bullous pemphigoid
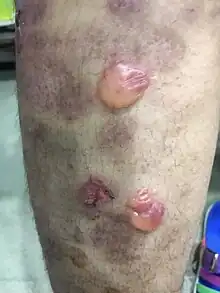
Bullous pemphigoid is a rare and chronic autoimmune disorder characterised by large sub-epidermal blisters called bullae, that predominantly involves the skin and less commonly the mucous membranes. It is the most common type of the pemphigoid group, representing 80% of sub-epidermal immunobullous cases.[9] It is more commonly known as cutaneous pemphigoid.
Presentation
Primary lesions of small and large blisters, known as vesicles and bullae, are found on the skin and sometimes on the mucous membranes. [10]
Non-bullous pemphigoid
In some patients, pemphigoid starts off with cutaneous manifestations of BP without bullae, as the only sign of the disease. Pruritic eczematous, papular, or urticaria-like skin lesions may also persist for weeks to months.[11]
Bullous phase
The bullous stage of BP shows vesicles and bulla, appearing on apparently normal or erythematous skin, predominantly at the flexural aspects of the extremities and the lower trunk.[11] Mucosal lesions, which typically are erosions of the oral mucosa, are present in 10 to 30 percent of patients.[12] Occasionally, the blister fluid becomes blood-tinged. The blisters are tense, about 1–4 cm in diameter, leaving eroded and crusted areas, together with urticarial and infiltrated papules and plaques in an annular or figurate pattern.[11][12]
Homology between bullous pemphigoid antigens in the skin and neuronal antigens in the central nervous system has been proposed as a cause for the observed link between bullous pemphigoid and neurologic disease, along with a genetic predisposition. Patients with bullous pemphigoid usually present with two or more other chronic diseases such as neurological disorders(dementia, Parkinson's disease, or stroke). However, further studies are necessary to explore the relationship between these disorders.[12]
Cause
The pathogenetic mechanism of blister formation is known, the trigger to the formation of the antibodies to the hemidesmosome antigens is still unknown.[13] Most of the bullous pemphigoid cases are due to autoantibodies (mostly IgG) directed at antigens (BP180 and BP230) arranged at the dermal-epidermal junction.[14] However, most commonly, drug can be one of the cause of bullous pemphigoid, such as thiazide diuretics, antibiotics (e.g., penicillins, vancomycin), nonsteroidal anti-inflammatory drugs (NSAIDs) and angiotensin-converting enzyme (ACE) inhibitors (e.g., captopril) and possibly angiotensin receptor blockers (ARBs, e.g., valsartan).[10]
The implicated drugs include penicillin derivatives, sulfasalazine, ibuprofen, phenacetin, enalapril, captopril, lisinopril, gabapentin, novoscabin, levobunolol ophthalmic solution, tetracoq, influenza, tetanus, and other vaccinations, a homeopathy regimen, nifedipine, 5-aminosalicylic acid, doxazosin, serratiopeptidase, losartan, cephalexin, bumetanide, fluoxetine, chloroquine, antipsychotic drugs, enoxaparin, ciprofloxacin, furosemide (frusemide), neuroleptics, penicillamine, gliptin plus metformin, intravenous iodine, etanercept, levofloxacin, and topical fluorouracil. Influenza vaccination does not appear to be an important trigger for bullous pemphigoid. Trauma, burns, lymphedema, phototherapy, and radiation have been implicated in a very small number of cases.[13]
Pathophysiology
The pathophysiology of bullous pemphigoid consists of two major components, which are immunologic and inflammatory. In the immunologic component, autoantibodies act against the hemidesmosomal bullous pemphigoid antigens BP230 (BPAg1) and BP 180(BPAg2 or type XVII collagen) which are located at the lamina lucida of the basement membrane zone. These antigens play an important role in the adhesion complexes that promote epithelial-stromal adhesion.[9] The predominant subclass of antibodies that acts against the antigens is IgG4. IgG1 and IgG2 antibodies are less frequently detected compared to IgG4 antibodies, while IgG3 antibodies are usually absent.[14] When the autoantibodies bind specifically to the target antigens, the complement system and mast cells are activated, thereby representing the inflammatory component. Inflammatory cells such as neutrophils and eosinophils are then attracted to the affected area. They are postulated to release proteolytic enzymes which degrade the hemidesmosomal proteins, resulting in blister formation.[9]
Other potential contributory factors including genetic factors, environmental exposures to infections and drugs as well as the phenomenon of epitope spreading are also known to cause bullous pemphigoid.[14]
Diagnosis
Diagnosis of bullous pemphigoid includes clinical assessment, skin biopsy for histopathology and direct immunofluorescence, indirect immunofluorescence and ELISA test. Among all, direct immunofluorescence is the gold standard for diagnosis of bullous pemphigoid.
Clinical assessment
For patients greater than 70 years old [15][16]
- Blistering skin disease characterized by the presence of tense blisters and erosions that occur without another identifiable cause and rarely on mucosa.
- Unexplained pruritus, pruritic eczematous eruptions, or urticarial plaques
Histopathology
Lesional tissue, preferably of an intact vesicle or the edge of an intact bulla is obtained using punch biopsy for Haemotoxylin and Eosin (H&E)staining.
Typical histopathologic findings include:[17][13]
- Sub-epidermal split with numerous eosinophils within the cleft.
- A superficial dermal inflammatory cell infiltrate of variable intensity with lymphocytes, eosinophils, and neutrophils.
- Eosinophlic spongiosis (Specifically in early lesion or may be seen in clinically erythematous skin surrounding the blister)
Direct immunofluorescence
Direct immunofluorescence (DIF) studies involves directly detecting tissue bound antibodies. Biopsy specimens for DIF should be taken from perilesional skin instead of lesional skin for H&E histopathologic evaluation. DIF specimens should be placed in Michel's solution or Zeuss transport media instead of formalin.
DIF of bullous pemphigoid will show the presence of fine, continuous and linear deposits of IgG and/or C3 along the epidermal basement membrane. Other classes of immunoglobulins such as IgM and IgA are present in approximately 20% of cases and usually are less intense. In some cases with the deposits of IgA, patient may have oral lesion. At early stages of the disease, only C3 may be present.[13]
Indirect immunofluorescence
Indirect immunofluorescence is used to detect circulating antibodies targeting the antigens at the basement membrane zone in patients with pemphigoid. In this procedure, patient's serum is collected and overlaid on salt-split normal human skin and incubated. Following this, the specimen will be stained for fluorescent detection of antibodies.
In bullous pemphigoid, circulating IgG targeting the basement membrane, mainly BP180 and BP230 hemidesmosomal proteins are detectable in 60-80% of patients. IgA and IgE classes can also be detected, but less frequently.[13]
Enzyme-linked immunosorbent assay (ELISA)
ELISA for bullous pemphigoid are commercially available to test for circulating Ig against NC16A domain of BP180 and BP230, known as bullous pemphigoid antigen 2 [BPAg2] and bullous pemphigoid antigen 1 [BPAg1] respectively. Antibodies to BP180NC16A domain is useful for the diagnosis of bullous pemphigoid as it has a sensitivity of 89% and specificity of 98%.[18]
Detection of BP180 and/or BP230 antibodies in serum does not give a confirmative diagnosis of bullous pemphigoid. A study has reported that 7% were tested positive for one or both autoantibodies in one series of 337 people without bullous pemphigoid.[19] ELISA findings should be correlated with DIF to reduce the risk of misdiagnosis.
Treatment
The treatment for bullous pemphigoid include:
1. Corticosteroids
i. Topical Corticosteroids
ii. Systemic corticosteroids
2. Glucocorticoid-sparing drugs
i. Immunosuppressive drugs
ii. Anti-inflammatory drugs
3. Biologic therapy
i. Intravenous immunoglobulin
ii. Rituximab
Among all, topical or systemic corticosteroids are considered as the first line therapy in controlling bullous pemphigoid. Other drugs and immunomodulatory therapies are often used as adjunct to minimize the adverse effect of long term use of corticosteroids and improve the healing of the disease.
There are several factors that have to be taken into account when choosing the therapies given to the patient: (a) patient's age (b) underlying disease such as hypertension, diabetes mellitus and other cardiovascular disease (c) side effect with the use of drugs (d) patient's ability to compliant to the therapy (d) severity and extent of disease (e) cost of drugs.
Corticosteroids
High potency topical corticosteroid is preferred as the first line treatment due to its efficacy and fewer systemic adverse effects when compared to systemic corticosteroids. Studies have shown that patients with extensive bullous pemphigoid (defined as >10 new bullae per day) treated with topical corticosteroids (Topical Clobetasol Propionate 0.05% cream) had better clinical outcomes than patients with extensive bullae pemphigoid who were treated with systemic glucocorticoid therapy (Prednisone).[20]
Systemic glucocorticoids can be used for patients when there are factors that make the use of topical corticosteroids not feasible, such as elderly patient inability to apply the cream on their own, cost or patient's own preference.
Topical Corticosteroids
Topical Clobetasol Propionate 0.05% cream is usually used and applied twice daily. A study by Joly et al. demonstrated that the use of 10 to 20g of Clobetasol Propionate per day for moderate disease and 20 to 30g per day for extensive disease until 15 days after disease control, then tapered to discontinuation over four months was as effective as the standard regime (40g per day tapered slowly over 12 months).[21]
Systemic corticosteroids
Prednisone is usually used to treat bullous pemphigoid. The dose varies between 0.2 and 0.5 mg/kg/day and will continue until active inflammation, new blister formation, pruritus has stopped for at least 2 weeks. The dose is then slowly tapered over the months. Initially, prednisolone can be reduced by relative large amounts (approximately 10 mg) and smaller amount (2.5–5 mg) subsequently. Should the patient develop flare up of the lesion, the dose should be increased to the previous level or higher and maintained longer before further, slower tapering.[22]
Glucocorticoid sparing drugs
For patients who require high dose of corticosteroids for clearing or maintenance, glucocorticoid sparing agents such as immunosuppressive drugs and anti-inflammatory drugs can be used as an adjunct therapy to reduce the systemic side effects of corticosteroids. Patients who have comorbidities and contraindications for corticosteroids may also consider these glucocorticoid sparing agents.
Immunosuppressant drug
Immunosuppressant drugs include azathioprine (1–3 mg/kg/day in two equally divided doses), mycophenolate mofetil (1000–3000 mg/day or 40 mg/kg/day in two divided doses), and methotrexate (10–15 mg/week).[22]
Anti-inflammatory drugs
Tetracycline antibiotics are often used in combination of nicotinamide to treat bullous pemphigoid.[23][24] For the administration of drugs, tetracycline is prescribed as 500 mg four times daily, doxycycline and minocycline as 100 mg twice daily and nicotinamide, 500 mg 4 times daily. Dapsone is also shown to be effective in treating bullous pemphigoid.[25] However, the efficacy of dapsone is limited. Dapsone is usually commenced at a low dose of 25 to 50 mg/day and increase by 25 mg every week until the condition improves. Maximum dose that can be prescribed is 250 mg/day.[22]
Biologic therapy
For refractory disease, biologic therapies such as intravenous immunoglobulin and Rituximab should be considered. [1,19,20][9][26][27]
Epidemiology
Bullous pemphigoid is primarily a disease of older adults and it rarely occurs in children. The vast majority of cases involved individuals between the ages of 60 and 80 years. Two European studies have also suggested the increased risk of bullous pemphigoid with advancing age.[28][29]
According to the results of several retrospective studies, there is an increasing incidence of bullous pemphigoid.[30][31][32] Bullous pemphigoid can be considered as the most common autoimmune blistering disease in Europe, while pemphigus may be more common in locations such as Thailand and Malaysia. It is reported that bullous pemphigoid has a slight female preponderance. However, the reasons for this are unknown.[14]
Mucous membrane pemphigoid
Mucous membrane pemphigoid (MMP), or cicatricial pemphigoid, is a rare, chronic, autoimmune sub-epidermal blistering disorder which predominantly involves the mucosae and has a tendency towards scarring of the affected areas.[11] Any mucous membrane can be involved, but the most commonly involved site is the oral mucosa, followed by conjunctiva, skin, pharynx, external genitalia, nasal mucosa, larynx, anus, and esophagus.[33] As MMP may lead to serious complications such as blindness and airway compression, early and aggressive treatment initiation may be needed.[34]
Presentation
Scarring is a common consequence of MMP that distinguishes this variant from mucosal involvement in bullous pemphigoid, which typically does not scar. Reticulated, white striations representing mucosal fibrosis often are present at sites of healed lesions, and functional limitations secondary to scarring may occur. As examples, MMP involving the ocular mucosa can lead to symblepharon, ankyloblepharon, and eventual blindness, and progressive laryngeal and tracheal involvement can result in asphyxiation.[14]
Oral disease
Most commonly affecting the mouth, including the buccal mucosa, gingiva, tongue, vermillion lips, and palate. Desquamative gingivitis is the most frequent manifestation.[35][34] The gingiva is erythematous, in which patients usually complaint of bleeding upon brushing.[34] Rupturing of oral vesiculobullous lesions leave clean, noninflamed, painless erosions. The vermilion border of the lips is spared, which is typical in pemphigus. Hoarseness due to laryngeal involvement can be seen in 8% of cases. A subset of patients have only oral disease, which has a relatively benign course compared with patients with oral cavity and other mucosae and skin involvement.[34]
Ocular disease
The eye is involved in 65% of cases. Initially presented with unilateral conjunctivitis (such as burning or excessive tearing), then fibrosis beneath the conjunctival epithelium.[35][36] Shrinkage of the conjunctiva leads to obliteration of the conjunctival sac.[35] Symblepharons are fibrous strands connecting the conjunctiva of the lid to the globe.[34] Besides, reduced tearing with erosion and neovascularization of the cornea leads to corneal opacification and perforation.[35] Scarring of the lid results in entropion (inward turning of the lid) and trichiasis (in-turning of the eyelashes).[37] These conditions ultimately lead to blindness in approximately 20% of cases. It is crucial to go for follow-up because relapse occurs in 22% of those who were in remission and not undergoing therapy.[35]
Other mucous membranes
Less common sites that might get involved are nasopharynx, esophagus, and urethra.[35] Nasopharyngeal involvement can lead to ulcerations of the septum and airway obstruction which might require tracheostomy.[35] Esophageal disease may present with ulcerations, dysphagia, odynophagia, and stenosis. Stenosis at urethra, vaginal orifice and rectal have also resulted from chronic inflammation and scarring.[35]
Skin disease
About 25% of patients have cutaneous lesions, with tense vesicles or bullae, mainly on the face, neck, and scalp. Healing of erosion is either with or without atrophic scars.[35] Cutaneous lesions of mucous membrane pemphigoid presents in 2 subtypes: (1)presents as generalized eruption of tense bullae without scarring (2) presents as localised blisters on an erythematous base, resulting in atrophic scarring.[36]
Mucous membrane pemphigoid is also associated with malignancy
Malignancy — MMP with antibodies directed against laminin 332 (previously known as laminin 5 and epiligrin) has been associated with an increased risk for internal malignancy. In a cohort of 35 patients with this type of pemphigoid (diagnosed with immunoprecipitation), 10 (29 percent) developed solid organ malignancies, 7 of which were diagnosed within 14 months after a diagnosis of MMP.[14] Occurrences of non-Hodgkin lymphoma and cutaneous T cell lymphoma have also been reported in individual patients with anti-laminin 332 MMP. The pathophysiologic relationship of this subtype of MMP to cancer is unknown. However, expression of laminin 332 has been detected in malignant cells, and laminin 332 appears to be capable of promoting tumor cell growth, invasion, and metastasis.[14]
The clinical manifestations of MMP in patients with laminin 332 antibodies are similar to the features of MMP with other antibody profiles.[14] Therefore, clinical examination cannot reliably distinguish anti-laminin 332 MMP from other forms of MMP. Additional studies are necessary to confirm the findings of a retrospective study of 154 patients with MPP that associated the detection of laminin 332 antibodies via a novel enzyme-linked immunosorbent assay (ELISA) with a greater likelihood for severe disease.
Since diagnostic laboratory testing for laminin 332 antibodies is not commercially available, suspicion for laminin 332 primarily is based upon immunofluorescence microscopy findings.[14] Although not exclusive to laminin 332 MPP, the detection of antibodies bound to the dermal side of basement membrane zone-split (salt-split) skin suggests the possibility of this diagnosis.
Until definitive testing for laminin 332 antibodies becomes available, we recommend that patients with MMP in whom serum indirect immunofluorescence (IIF) studies reveal antibodies bound to the dermal side of basement membrane zone-split skin undergo age and gender appropriate cancer screening.[14] Additional evaluation for malignancy should be performed as indicated based upon a review of symptoms, physical examination, and the results of age-appropriate screening.
Cause
Autoantibodies targeted to components of the basement membrane zone have been identified as pathogenic in mucous membrane pemphigoid. Antigens include 180-kD bullous pemphigoid antigen (BP180), laminin 332, beta-4-integrin, and other antigens that are not fully discovered are identified against the basement membrane.[34] Complication of D- penicillamine therapy may trigger and causes mucous membrane pemphigoid. It also occurs after acute severe ocular inflammation in patients with Stevens-Johnson syndrome.[38]
Pathophysiology
Autoantibodies target the basement membrane zone proteins which are responsible to promote adhesion within the basement membrane zone of the mucosa and the skin. The major basement membrane zone proteins identified include :
- C-terminus of BP180
- BP230
- Laminin 332(also known as laminin 5 or epiligrin)
- Alpha-6-beta-4 integrin Type VII collagen.
In contrast to the target of the N-terminus of BP180 that is located in the hemidesmosomes and upper lamina lucida in bullous pemphigoid, the target antigen in MMP is the C-terminus of BP180 which is located in the lower lamina lucida and lamina densa. This results in a deeper separation that is more likely to scar as compared to a more superficial blister that is unlikely to scar in bullous pemphigoid.
Antibodies to the beta-4 integrin subunit of alpha-6-beta-4 integrin is shown to be associated with ocular disease while oral involvement is suggested to be linked with antibody reaction towards the alpha-6 subunit. Besides, MMP with antibody reaction against laminin 332 has an association with an increased risk for internal malignancy.
Similar to bullous pemphigoid, other factors such as genetic factors, environmental exposures and the phenomenon of epitope spreading potentially result in MMP. Multiple studies have also reported an association of HLA-DQB1*0301 with MPP.[14][39][40][41][42]
Diagnosis
Diagnosis of bullous pemphigoid includes clinical assessment, skin biopsy for histopathology and direct immunofluorescence, indirect immunofluorescence and ELISA test. Among all, direct immunofluorescence is the gold standard for diagnosis of mucous membrane pemphigoid.
Clinical assessment
- Presence of tense blisters and erosions that occur on skin without another identifiable cause.
- Desquamative gingivitis or mucositis involving oral, ocular, nasal, genital, anal, pharyngeal, laryngeal, and/or esophageal mucosae
- Presence of pruritic eczematous eruptions, or urticarial plaques without identifiable cause.
- Patient's age over 60 years old.
Histopathology
Lesional tissue, preferably of an intact vesicle or the edge of an intact bulla is obtained using punch biopsy for Haemotoxylin and Eosin (H&E) staining.
The findings are sub-epidermal blister with dermal infiltrated with lymphocytes, neutrophils and eosinophils. Additional findings include sub-epidermal fibrosis which is consistent with the scarring nature of mucous membrane pemphigoid in older lesions and plasma cell infiltration.[37][36]
Direct immunofluorescence studies
Direct immunofluorescence (DIF) studies involves directly detecting tissue bound antibodies. Biopsy specimens for DIF should be taken from perilesional skin instead of lesional skin for H&E histopathologic evaluation. Linear band of IgG and C3 deposits are found along the basement membrane. Occasionally, linear deposition of IgA at the basement membrane zone can also be seen.[43][44] Multiple and repeated biopsies increase the sensitivity of DIF studies to diagnose MMP.
Indirect immunofluorescence
Indirect immunofluorescence is used to detect circulating antibodies targeting the antigens at the basement membrane zone in patients with pemphigoid. In early studies using routine techniques, only one third of patient with MPP were being tested positive. Circulating IgG and IgA antibodies are found in patient's serum. To increase the likelihood of detecting circulating antibodies, human basement membrane zone-split skin and/or concentrated serum should be used.[37][45][46]
Antigen-specific serologic testing
Autoantibodies directed against a variety of antigens, including BP180, BP230, laminin 332, and type VII collagen may be detected.[47][48][49] However, this test could not be used as the only diagnostic tool for testing as ELISA testing has limited sensitivity.
Treatment
The factors that determine the type of therapy used for mucous membrane pemphigoid are: [1] site(s) of involvement, [2] severity of disease, [3] rate of progression.
Oral mucosa is the most common site being affected in mucous membrane pemphigoid.
For the mild oral mucosa lesion, high potency topical steroids such as 0.05% Clobetasol propionate is used. Patients are instructed to apply the ointment or gel 2-3 times a day after drying the oral mucosa to enhance the adherence of mediation to oral mucosa. Patients are instructed to avoid drinking or eating for at least 30 minutes after application. Dental tray can also be fabricated to help in the application of topical steroids to lesional sites under occlusion for patients with gingival involvement. Furthermore, topical tacrolimus, a calcineurin inhibitor, has also shown to be effective to control the disease, including some patients who failed to respond well to topical corticosteroids. Topical tacrolimus 0.1% ointment is applied two to three times a day and tapered after improvement in healing of pemphigoid. Another method is to use intralesional corticosteroids (Triamcinolone acetonide, dilution of 5 to 10 mg/ml; repeated every 2–4 weeks). Intralesional therapy is used when the patient does not respond to local therapies.
For moderate to severe disease (including the ones involving ocular, nasopharyngeal, or anogenital mucosa) and patient who did not respond to local therapy adequately, systemic agents should be used. Systemic corticosteroids and dapsone are used in such cases. The dose of dapsone ranges from 50 to 200 mg daily. Dapsone is shown to be effective in treating mucous membrane pemphigoid that does not respond to systemic corticosteroids.[37] Whereas for systemic corticosteroids, 0.25 to 0.5 mg/kg of prednisolone is prescribed per day (twice-daily dosage is used during the acute stage and change to a single daily morning dose after new blister formation stops). Thereafter, the dosage of prednisolone is slowly tapered over the months in combination with topical therapy or glucocorticoid-sparing agent (e.g., dapsone, azathioprine).
Patients with severe mucous membrane pemphigoid that cannot be controlled by the intervention above and would need aggressive immunosuppressive regimens and biologic therapies to control the lesions.[37] Azathioprine or Cyclosphosphamide are the choices of immunosuppressive drugs that can be used. Sometimes, immunosuppressive agents and prednisolone can be combined if dapsone fails to improve the condition. Lastly, in patients who do not respond to the conventional therapy, rituximab may be an option.[35]
There is insufficient evidence that cyclophosphamide combined with corticosteriods are effective in treating mucous membrane pemphigoid.[50]
Other than that, oral hygiene instructions should be given to patients as oral care is a critical part in treating mucous membrane pemphigoid.[51] Before meals, patients are advised to rinse with hydrogen peroxide (diluted with water to a concentration of 1:4 or 1:6) and diphenhydramine to reduce the pain. Patient would then rinse with hydrogen peroxide to remove food particles and debris and later rinse with dexamethasone for anti-inflammatory effect. Hydrogen peroxide, elixir of dexamethasone and elixir of diphenhydramine are each diluted with water to a concentration of 1:4 or 1:6 and are instructed not to swallow in the end.[35]
Epidemiology
MMP mainly affect the elderly population of ages between 60 and 80 years and rarely children. Women are affected twice as frequently than in men.[52] There is no known racial or geographic predilection, but several studies have suggested that there is an association of specific immunogenetic haplotype HLA-DQB1*0301 with MMP.[11][39][40][41][42]
References
- "pemphigoid" at Dorland's Medical Dictionary
- Pemphigoid at the U.S. National Library of Medicine Medical Subject Headings (MeSH)
- "British Association of Dermatologists - Patient Information Leaflets (PILs)". www.bad.org.uk. Archived from the original on 11 June 2020. Retrieved 11 June 2020.
- Cicatricial Pemphigoid at eMedicine
- Bullous Pemphigoid at eMedicine
- Pemphigoid Gestationis at eMedicine
- Provost TT, Flynn JA (2001). Cutaneous medicine: cutaneous manifestations of systemic disease. PMPH-USA. pp. 209–. ISBN 978-1-55009-100-7. Retrieved 25 June 2010.
- He Y, Shimoda M, Ono Y, Villalobos IB, Mitra A, Konia T, et al. (June 2015). "Persistence of Autoreactive IgA-Secreting B Cells Despite Multiple Immunosuppressive Medications Including Rituximab". JAMA Dermatology. 151 (6): 646–50. doi:10.1001/jamadermatol.2015.59. PMID 25901938.
- Baigrie D, Nookala V (2020). "Bullous Pemphigoid". StatPearls. StatPearls Publishing. PMID 30570995. Retrieved 2019-01-19.
- Mirowski GW, Leblanc J, Mark LA (2016). "Oral disease and oral-cutaneous manifestations of gastrointestinal and liver disease". In Feldman M (ed.). Sleisenger & Fordtran's Gastrointestinal and Liver Disease (10th ed.). Philadelphia, Pa: Saunders Elsevier. pp. 377–396.
- Bernard P, Borradori L (2018). "Pemphigoid Group". In Bolognia JL (ed.). Dermatology (4th ed.). Philadelphia, Pa: Elsevier Limited. pp. 510–526.
- Chow YW, Pietranico R, Mukerji A (October 1975). "Studies of oxygen binding energy to hemoglobin molecule". Biochemical and Biophysical Research Communications. 66 (4): 1424–31. doi:10.1016/0006-291X(75)90518-5. PMID 6.
- Patterson JW, Hosler GA, Weedon D (2016). Weedon's Skin Pathology (4th ed.). London: Churchill Livingstone Elsevier. ISBN 978-0-7020-5183-8.
- Leiferman KM. Zone JJ, Ofori AO (eds.). "Epidemiology and pathogenesis of bullous pemphigoid and mucous membrane pemphigoid". UpToDate. Wolters Kluwer. Retrieved 2019-01-17.
- Vaillant L, Bernard P, Joly P, Prost C, Labeille B, Bedane C, et al. (September 1998). "Evaluation of clinical criteria for diagnosis of bullous pemphigoid. French Bullous Study Group". Archives of Dermatology. 134 (9): 1075–80. doi:10.1001/archderm.134.9.1075. PMID 9762017.
- Kershenovich R, Hodak E, Mimouni D (2014-04-01). "Diagnosis and classification of pemphigus and bullous pemphigoid". Autoimmunity Reviews. Diagnostic criteria in Autoimmune diseases. 13 (4–5): 477–81. doi:10.1016/j.autrev.2014.01.011. PMID 24424192.
- Schmidt E, della Torre R, Borradori L (July 2011). "Clinical features and practical diagnosis of bullous pemphigoid". Dermatologic Clinics. 29 (3): 427–38, viii–ix. doi:10.1016/j.det.2011.03.010. PMID 21605808.
- Samuel M, Pixley RA, Villanueva MA, Colman RW, Villanueva GB (September 1992). "Human factor XII (Hageman factor) autoactivation by dextran sulfate. Circular dichroism, fluorescence, and ultraviolet difference spectroscopic studies". The Journal of Biological Chemistry. 267 (27): 19691–7. doi:10.1016/S0021-9258(18)41830-3. PMID 1527088.
- Wieland CN, Comfere NI, Gibson LE, Weaver AL, Krause PK, Murray JA (January 2010). "Anti-bullous pemphigoid 180 and 230 antibodies in a sample of unaffected subjects". Archives of Dermatology. 146 (1): 21–5. doi:10.1001/archdermatol.2009.331. PMID 20083688.
- Joly P, Roujeau JC, Benichou J, Picard C, Dreno B, Delaporte E, et al. (January 2002). "A comparison of oral and topical corticosteroids in patients with bullous pemphigoid". The New England Journal of Medicine. 346 (5): 321–7. doi:10.1056/NEJMoa011592. PMID 11821508.
- Joly P, Roujeau JC, Benichou J, Delaporte E, D'Incan M, Dreno B, et al. (July 2009). "A comparison of two regimens of topical corticosteroids in the treatment of patients with bullous pemphigoid: a multicenter randomized study". The Journal of Investigative Dermatology. 129 (7): 1681–7. doi:10.1038/jid.2008.412. PMID 19177141.
- Kellerman RD, Rakel D, eds. (2020). "Bulbous Disease: Method of Diya F. Mutasim". Conn's current therapy 2020 (20th ed.). Philadelphia: Elsevier. p. 228. ISBN 978-0-323-71184-5.
- Berk MA, Lorincz AL (June 1986). "The treatment of bullous pemphigoid with tetracycline and niacinamide. A preliminary report". Archives of Dermatology. 122 (6): 670–4. doi:10.1001/archderm.1986.01660180076019. PMID 2940979.
- Thomas I, Khorenian S, Arbesfeld DM (January 1993). "Treatment of generalized bullous pemphigoid with oral tetracycline". Journal of the American Academy of Dermatology. 28 (1): 74–7. doi:10.1016/0190-9622(93)70013-J. PMID 8425974.
- Bouscarat F, Chosidow O, Picard-Dahan C, Sakiz V, Crickx B, Prost C, et al. (April 1996). "Treatment of bullous pemphigoid with dapsone: retrospective study of thirty-six cases". Journal of the American Academy of Dermatology. 34 (4): 683–4. doi:10.1016/S0190-9622(96)80085-5. PMID 8601662.
- Czernik A, Toosi S, Bystryn JC, Grando SA (February 2012). "Intravenous immunoglobulin in the treatment of autoimmune bullous dermatoses: an update". Autoimmunity. 45 (1): 111–8. doi:10.3109/08916934.2011.606452. PMID 21923613. S2CID 19942412.
- Kasperkiewicz M, Shimanovich I, Ludwig RJ, Rose C, Zillikens D, Schmidt E (September 2011). "Rituximab for treatment-refractory pemphigus and pemphigoid: a case series of 17 patients". Journal of the American Academy of Dermatology. 65 (3): 552–558. doi:10.1016/j.jaad.2010.07.032. PMID 21641080.
- Marazza G, Pham HC, Schärer L, Pedrazzetti PP, Hunziker T, Trüeb RM, et al. (October 2009). "Incidence of bullous pemphigoid and pemphigus in Switzerland: a 2-year prospective study". The British Journal of Dermatology. 161 (4): 861–8. doi:10.1111/j.1365-2133.2009.09300.x. PMID 19566661. S2CID 30118554.
- Bertram F, Bröcker EB, Zillikens D, Schmidt E (May 2009). "Prospective analysis of the incidence of autoimmune bullous disorders in Lower Franconia, Germany". Journal of the German Society of Dermatology. 7 (5): 434–40. doi:10.1111/j.1610-0387.2008.06976.x. PMID 19170813. S2CID 25769559.
- Joly P, Baricault S, Sparsa A, Bernard P, Bédane C, Duvert-Lehembre S, et al. (August 2012). "Incidence and mortality of bullous pemphigoid in France". The Journal of Investigative Dermatology. 132 (8): 1998–2004. doi:10.1038/jid.2012.35. PMID 22418872.
- Försti AK, Jokelainen J, Timonen M, Tasanen K (November 2014). "Increasing incidence of bullous pemphigoid in Northern Finland: a retrospective database study in Oulu University Hospital". The British Journal of Dermatology. 171 (5): 1223–6. doi:10.1111/bjd.13189. PMID 24934834. S2CID 207071905.
- Brick KE, Weaver CH, Lohse CM, Pittelkow MR, Lehman JS, Camilleri MJ, et al. (July 2014). "Incidence of bullous pemphigoid and mortality of patients with bullous pemphigoid in Olmsted County, Minnesota, 1960 through 2009". Journal of the American Academy of Dermatology. 71 (1): 92–9. doi:10.1016/j.jaad.2014.02.030. PMC 4324601. PMID 24704091.
- Habif TP, Campbell JL, Chapman MS, Dinulos JG, Zug KA (2011), "Vesicular and bullous diseases", Skin Disease, Elsevier, pp. 366–377, doi:10.1016/b978-0-323-07700-2.00013-5, ISBN 9780323077002
- Tolaymat L, Hall MR (2020). "Cicatricial Pemphigoid". StatPearls. StatPearls Publishing. PMID 30252376. Retrieved 2019-01-19.
- Mascaró JM (2010). "Other Vesiculobullous Diseases". Sleisenger & Fordtran's Gastrointestinal and Liver Disease (9th ed.). Philadelphia, Pa: Saunders Elsevier.
- Domloge-Hultsch N, Gammon WR, Briggaman RA, Gil SG, Carter WG, Yancey KB (October 1992). "Epiligrin, the major human keratinocyte integrin ligand, is a target in both an acquired autoimmune and an inherited subepidermal blistering skin disease". The Journal of Clinical Investigation. 90 (4): 1628–33. doi:10.1172/JCI116033. PMC 443212. PMID 1401088.
- Fleming TE, Korman NJ (October 2000). "Cicatricial pemphigoid". Journal of the American Academy of Dermatology. 43 (4): 571–91, quiz 591–4. doi:10.1067/mjd.2000.107248. PMID 11004612.
- Hamodat M. "Cicatricial pemphigoid". www.pathologyoutlines.com. Retrieved 2019-01-17.
- Chan LS, Hammerberg C, Cooper KD (February 1997). "Significantly increased occurrence of HLA-DQB1*0301 allele in patients with ocular cicatricial pemphigoid". The Journal of Investigative Dermatology. 108 (2): 129–32. doi:10.1111/1523-1747.ep12332352. PMID 9008223.
- Delgado JC, Turbay D, Yunis EJ, Yunis JJ, Morton ED, Bhol K, et al. (August 1996). "A common major histocompatibility complex class II allele HLA-DQB1* 0301 is present in clinical variants of pemphigoid". Proceedings of the National Academy of Sciences of the United States of America. 93 (16): 8569–71. Bibcode:1996PNAS...93.8569D. doi:10.1073/pnas.93.16.8569. PMC 38713. PMID 8710911.
- Setterfield J, Theron J, Vaughan RW, Welsh KI, Mallon E, Wojnarowska F, et al. (September 2001). "Mucous membrane pemphigoid: HLA-DQB1*0301 is associated with all clinical sites of involvement and may be linked to antibasement membrane IgG production". The British Journal of Dermatology. 145 (3): 406–14. doi:10.1046/j.1365-2133.2001.04380.x. PMID 11531829.
- Yunis JJ, Mobini N, Yunis EJ, Alper CA, Deulofeut R, Rodriguez A, et al. (August 1994). "Common major histocompatibility complex class II markers in clinical variants of cicatricial pemphigoid". Proceedings of the National Academy of Sciences of the United States of America. 91 (16): 7747–51. Bibcode:1994PNAS...91.7747Y. doi:10.1073/pnas.91.16.7747. PMC 44479. PMID 8052655.
- Bean SF, Waisman M, Michel B, Thomas CI, Knox JM, Levine M (August 1972). "Cicatricial pemphigoid. Immunofluorescent studies". Archives of Dermatology. 106 (2): 195–9. doi:10.1001/archderm.1972.01620110031007. PMID 4558699.
- Leonard JN, Wright P, Williams DM, Gilkes JJ, Haffenden GP, McMinn RM, Fry L (March 1984). "The relationship between linear IgA disease and benign mucous membrane pemphigoid". The British Journal of Dermatology. 110 (3): 307–14. doi:10.1111/j.1365-2133.1984.tb04636.x. PMID 6365149. S2CID 8889595.
- Kelly SE, Wojnarowska F (January 1988). "The use of chemically split tissue in the detection of circulating anti-basement membrane zone antibodies in bullous pemphigoid and cicatricial pemphigoid". The British Journal of Dermatology. 118 (1): 31–40. doi:10.1111/j.1365-2133.1988.tb01747.x. PMID 3277659. S2CID 43832261.
- Setterfield J, Shirlaw PJ, Kerr-Muir M, Neill S, Bhogal BS, Morgan P, et al. (April 1998). "Mucous membrane pemphigoid: a dual circulating antibody response with IgG and IgA signifies a more severe and persistent disease". The British Journal of Dermatology. 138 (4): 602–10. doi:10.1046/j.1365-2133.1998.02168.x. PMID 9640363. S2CID 20114355.
- Bernard P, Antonicelli F, Bedane C, Joly P, Le Roux-Villet C, Duvert-Lehembre S, et al. (May 2013). "Prevalence and clinical significance of anti-laminin 332 autoantibodies detected by a novel enzyme-linked immunosorbent assay in mucous membrane pemphigoid". JAMA Dermatology. 149 (5): 533–40. doi:10.1001/jamadermatol.2013.1434. PMID 23426192.
- Yasukochi A, Teye K, Ishii N, Hashimoto T (August 2016). "Clinical and Immunological Studies of 332 Japanese Patients Tentatively Diagnosed as Anti-BP180-type Mucous Membrane Pemphigoid: A Novel BP180 C-terminal Domain Enzyme-linked Immunosorbent Assay". Acta Dermato-Venereologica. 96 (6): 762–7. doi:10.2340/00015555-2407. PMID 26984589.
- Sezin T, Egozi E, Hillou W, Avitan-Hersh E, Bergman R (July 2013). "Anti-laminin-332 mucous membrane pemphigoid developing after a diphtheria tetanus vaccination". JAMA Dermatology. 149 (7): 858–62. doi:10.1001/jamadermatol.2013.741. PMID 23700098.
- Kirtschig G, Murrell D, Wojnarowska F, Khumalo N, et al. (Cochrane Skin Group) (2003-01-20). "Interventions for mucous membrane pemphigoid and epidermolysis bullosa acquisita". The Cochrane Database of Systematic Reviews. 2015 (1): CD004056. doi:10.1002/14651858.CD004056. PMC 8406492. PMID 12535507.
- Knudson RM, Kalaaji AN, Bruce AJ (May 2010). "The management of mucous membrane pemphigoid and pemphigus". Dermatologic Therapy. 23 (3): 268–80. doi:10.1111/j.1529-8019.2010.01323.x. PMID 20597945. S2CID 205694155.
- Xu HH, Werth VP, Parisi E, Sollecito TP (October 2013). "Mucous membrane pemphigoid". Dental Clinics of North America. 57 (4): 611–30. doi:10.1016/j.cden.2013.07.003. PMC 3928007. PMID 24034069.