Phagocyte
Phagocytes are cells that protect the body by ingesting harmful foreign particles, bacteria, and dead or dying cells. Their name comes from the Greek phagein, "to eat" or "devour", and "-cyte", the suffix in biology denoting "cell", from the Greek kutos, "hollow vessel".[1] They are essential for fighting infections and for subsequent immunity.[2] Phagocytes are important throughout the animal kingdom[3] and are highly developed within vertebrates.[4] One litre of human blood contains about six billion phagocytes.[5] They were discovered in 1882 by Ilya Ilyich Mechnikov while he was studying starfish larvae.[6] Mechnikov was awarded the 1908 Nobel Prize in Physiology or Medicine for his discovery.[7] Phagocytes occur in many species; some amoebae behave like macrophage phagocytes, which suggests that phagocytes appeared early in the evolution of life.[8]
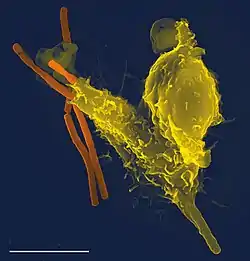
Phagocytes of humans and other animals are called "professional" or "non-professional" depending on how effective they are at phagocytosis.[9] The professional phagocytes include many types of white blood cells (such as neutrophils, monocytes, macrophages, mast cells, and dendritic cells).[10] The main difference between professional and non-professional phagocytes is that the professional phagocytes have molecules called receptors on their surfaces that can detect harmful objects, such as bacteria, that are not normally found in the body. Non-professional phagocytes do not have efficient phagocytic receptors, such as those for opsonins.[11] Phagocytes are crucial in fighting infections, as well as in maintaining healthy tissues by removing dead and dying cells that have reached the end of their lifespan.[12]
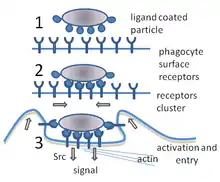
During an infection, chemical signals attract phagocytes to places where the pathogen has invaded the body. These chemicals may come from bacteria or from other phagocytes already present. The phagocytes move by a method called chemotaxis. When phagocytes come into contact with bacteria, the receptors on the phagocyte's surface will bind to them. This binding will lead to the engulfing of the bacteria by the phagocyte.[13] Some phagocytes kill the ingested pathogen with oxidants and nitric oxide.[14] After phagocytosis, macrophages and dendritic cells can also participate in antigen presentation, a process in which a phagocyte moves parts of the ingested material back to its surface. This material is then displayed to other cells of the immune system. Some phagocytes then travel to the body's lymph nodes and display the material to white blood cells called lymphocytes. This process is important in building immunity,[15] and many pathogens have evolved methods to evade attacks by phagocytes.[2]
History

The Russian zoologist Ilya Ilyich Mechnikov (1845–1916) first recognized that specialized cells were involved in defense against microbial infections.[16] In 1882, he studied motile (freely moving) cells in the larvae of starfishes, believing they were important to the animals' immune defenses. To test his idea, he inserted small thorns from a tangerine tree into the larvae. After a few hours he noticed that the motile cells had surrounded the thorns.[16] Mechnikov traveled to Vienna and shared his ideas with Carl Friedrich Claus who suggested the name "phagocyte" (from the Greek words phagein, meaning "to eat or devour", and kutos, meaning "hollow vessel"[1]) for the cells that Mechnikov had observed.[17]
A year later, Mechnikov studied a fresh water crustacean called Daphnia, a tiny transparent animal that can be examined directly under a microscope. He discovered that fungal spores that attacked the animal were destroyed by phagocytes. He went on to extend his observations to the white blood cells of mammals and discovered that the bacterium Bacillus anthracis could be engulfed and killed by phagocytes, a process that he called phagocytosis.[18] Mechnikov proposed that phagocytes were a primary defense against invading organisms.[16]
In 1903, Almroth Wright discovered that phagocytosis was reinforced by specific antibodies that he called opsonins, from the Greek opson, "a dressing or relish".[19] Mechnikov was awarded (jointly with Paul Ehrlich) the 1908 Nobel Prize in Physiology or Medicine for his work on phagocytes and phagocytosis.[7]
Although the importance of these discoveries slowly gained acceptance during the early twentieth century, the intricate relationships between phagocytes and all the other components of the immune system were not known until the 1980s.[20]
Phagocytosis
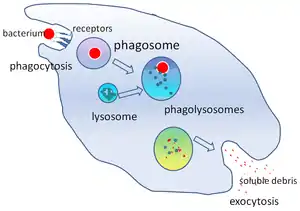
Phagocytosis is the process of taking in particles such as bacteria, invasive fungi, parasites, dead host cells, and cellular and foreign debris by a cell.[22] It involves a chain of molecular processes.[23][24] Phagocytosis occurs after the foreign body, a bacterial cell, for example, has bound to molecules called "receptors" that are on the surface of the phagocyte. The phagocyte then stretches itself around the bacterium and engulfs it. Phagocytosis of bacteria by human neutrophils takes on average nine minutes.[25] Once inside this phagocyte, the bacterium is trapped in a compartment called a phagosome. Within one minute the phagosome merges with either a lysosome or a granule to form a phagolysosome. The bacterium is then subjected to an overwhelming array of killing mechanisms[26] and is dead a few minutes later.[25] Dendritic cells and macrophages are not so fast, and phagocytosis can take many hours in these cells. Macrophages are slow and untidy eaters; they engulf huge quantities of material and frequently release some undigested back into the tissues. This debris serves as a signal to recruit more phagocytes from the blood.[27] Phagocytes have voracious appetites; scientists have even fed macrophages with iron filings and then used a small magnet to separate them from other cells.[28]
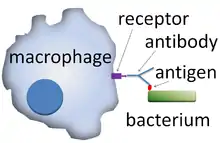
A phagocyte has many types of receptors on its surface that are used to bind material.[2] They include opsonin receptors, scavenger receptors, and Toll-like receptors. Opsonin receptors increase the phagocytosis of bacteria that have been coated with immunoglobulin G (IgG) antibodies or with complement. "Complement" is the name given to a complex series of protein molecules found in the blood that destroy cells or mark them for destruction.[29] Scavenger receptors bind to a large range of molecules on the surface of bacterial cells, and Toll-like receptors—so called because of their similarity to well-studied receptors in fruit flies that are encoded by the Toll gene—bind to more specific molecules including foreign DNA and RNA.[30] Binding to Toll-like receptors increases phagocytosis and causes the phagocyte to release a group of hormones that cause inflammation.[2]
Methods of killing
The killing of microbes is a critical function of phagocytes that is performed either within the phagocyte (intracellular killing) or outside of the phagocyte (extracellular killing).[31]
Oxygen-dependent intracellular
When a phagocyte ingests bacteria (or any material), its oxygen consumption increases. The increase in oxygen consumption, called a respiratory burst, produces reactive oxygen-containing molecules that are anti-microbial.[32] The oxygen compounds are toxic to both the invader and the cell itself, so they are kept in compartments inside the cell. This method of killing invading microbes by using the reactive oxygen-containing molecules is referred to as oxygen-dependent intracellular killing, of which there are two types.[14]
The first type is the oxygen-dependent production of a superoxide,[2] which is an oxygen-rich bacteria-killing substance.[33] The superoxide is converted to hydrogen peroxide and singlet oxygen by an enzyme called superoxide dismutase. Superoxides also react with the hydrogen peroxide to produce hydroxyl radicals, which assist in killing the invading microbe.[2]
The second type involves the use of the enzyme myeloperoxidase from neutrophil granules.[34] When granules fuse with a phagosome, myeloperoxidase is released into the phagolysosome, and this enzyme uses hydrogen peroxide and chlorine to create hypochlorite, a substance used in domestic bleach. Hypochlorite is extremely toxic to bacteria.[2] Myeloperoxidase contains a heme pigment, which accounts for the green color of secretions rich in neutrophils, such as pus and infected sputum.[35]
Oxygen-independent intracellular

Phagocytes can also kill microbes by oxygen-independent methods, but these are not as effective as the oxygen-dependent ones. There are four main types. The first uses electrically charged proteins that damage the bacterium's membrane. The second type uses lysozymes; these enzymes break down the bacterial cell wall. The third type uses lactoferrins, which are present in neutrophil granules and remove essential iron from bacteria.[36] The fourth type uses proteases and hydrolytic enzymes; these enzymes are used to digest the proteins of destroyed bacteria.[37]
Extracellular
Interferon-gamma—which was once called macrophage activating factor—stimulates macrophages to produce nitric oxide. The source of interferon-gamma can be CD4+ T cells, CD8+ T cells, natural killer cells, B cells, natural killer T cells, monocytes, other macrophages, or dendritic cells.[38] Nitric oxide is then released from the macrophage and, because of its toxicity, kills microbes near the macrophage.[2] Activated macrophages produce and secrete tumor necrosis factor. This cytokine—a class of signaling molecule[39]—kills cancer cells and cells infected by viruses, and helps to activate the other cells of the immune system.[40]
In some diseases, e.g., the rare chronic granulomatous disease, the efficiency of phagocytes is impaired, and recurrent bacterial infections are a problem.[41] In this disease there is an abnormality affecting different elements of oxygen-dependent killing. Other rare congenital abnormalities, such as Chédiak–Higashi syndrome, are also associated with defective killing of ingested microbes.[42]
Viruses
Viruses can reproduce only inside cells, and they can gain entry by using many of the receptors involved in immunity. Once inside the cell, viruses use the cell's biological machinery to their own advantage, forcing the cell to make hundreds of identical copies of themselves. Although phagocytes and other components of the innate immune system can, to a limited extent, control viruses, once a virus is inside a cell the adaptive immune responses, particularly the lymphocytes, are more important for defense.[43] At the sites of viral infections, lymphocytes often vastly outnumber all the other cells of the immune system; this is common in viral meningitis.[44] Virus-infected cells that have been killed by lymphocytes are cleared from the body by phagocytes.[45]
Role in apoptosis
In an animal, cells are constantly dying. A balance between cell division and cell death keeps the number of cells relatively constant in adults.[12] There are two different ways a cell can die: by necrosis or by apoptosis. In contrast to necrosis, which often results from disease or trauma, apoptosis—or programmed cell death—is a normal healthy function of cells. The body has to rid itself of millions of dead or dying cells every day, and phagocytes play a crucial role in this process.[46]
Dying cells that undergo the final stages of apoptosis[47] display molecules, such as phosphatidylserine, on their cell surface to attract phagocytes.[48] Phosphatidylserine is normally found on the cytosolic surface of the plasma membrane, but is redistributed during apoptosis to the extracellular surface by a protein known as scramblase.[49][50] These molecules mark the cell for phagocytosis by cells that possess the appropriate receptors, such as macrophages.[51] The removal of dying cells by phagocytes occurs in an orderly manner without eliciting an inflammatory response and is an important function of phagocytes.[52]
Interactions with other cells
Phagocytes are usually not bound to any particular organ but move through the body interacting with the other phagocytic and non-phagocytic cells of the immune system. They can communicate with other cells by producing chemicals called cytokines, which recruit other phagocytes to the site of infections or stimulate dormant lymphocytes.[53] Phagocytes form part of the innate immune system, which animals, including humans, are born with. Innate immunity is very effective but non-specific in that it does not discriminate between different sorts of invaders. On the other hand, the adaptive immune system of jawed vertebrates—the basis of acquired immunity—is highly specialized and can protect against almost any type of invader.[54] The adaptive immune system is not dependent on phagocytes but lymphocytes, which produce protective proteins called antibodies, which tag invaders for destruction and prevent viruses from infecting cells.[55] Phagocytes, in particular dendritic cells and macrophages, stimulate lymphocytes to produce antibodies by an important process called antigen presentation.[56]
Antigen presentation

Antigen presentation is a process in which some phagocytes move parts of engulfed materials back to the surface of their cells and "present" them to other cells of the immune system.[57] There are two "professional" antigen-presenting cells: macrophages and dendritic cells.[58] After engulfment, foreign proteins (the antigens) are broken down into peptides inside dendritic cells and macrophages. These peptides are then bound to the cell's major histocompatibility complex (MHC) glycoproteins, which carry the peptides back to the phagocyte's surface where they can be "presented" to lymphocytes.[15] Mature macrophages do not travel far from the site of infection, but dendritic cells can reach the body's lymph nodes, where there are millions of lymphocytes.[59] This enhances immunity because the lymphocytes respond to the antigens presented by the dendritic cells just as they would at the site of the original infection.[60] But dendritic cells can also destroy or pacify lymphocytes if they recognize components of the host body; this is necessary to prevent autoimmune reactions. This process is called tolerance.[61]
Immunological tolerance
Dendritic cells also promote immunological tolerance,[62] which stops the body from attacking itself. The first type of tolerance is central tolerance, that occurs in the thymus. T cells that bind (via their T cell receptor) to self antigen (presented by dendritic cells on MHC molecules) too strongly are induced to die. The second type of immunological tolerance is peripheral tolerance. Some self reactive T cells escape the thymus for a number of reasons, mainly due to the lack of expression of some self antigens in the thymus. Another type of T cell; T regulatory cells can down regulate self reactive T cells in the periphery.[63] When immunological tolerance fails, autoimmune diseases can follow.[64]
Professional phagocytes
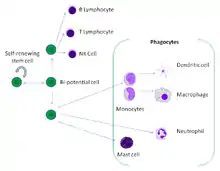
Phagocytes of humans and other jawed vertebrates are divided into "professional" and "non-professional" groups based on the efficiency with which they participate in phagocytosis.[9] The professional phagocytes are myeloid cells, which includes monocytes, macrophages, neutrophils, tissue dendritic cells and mast cells.[10] One litre of human blood contains about six billion phagocytes.[5]
Activation
All phagocytes, and especially macrophages, exist in degrees of readiness. Macrophages are usually relatively dormant in the tissues and proliferate slowly. In this semi-resting state, they clear away dead host cells and other non-infectious debris and rarely take part in antigen presentation. But, during an infection, they receive chemical signals—usually interferon gamma—which increases their production of MHC II molecules and which prepares them for presenting antigens. In this state, macrophages are good antigen presenters and killers. If they receive a signal directly from an invader, they become "hyperactivated", stop proliferating, and concentrate on killing. Their size and rate of phagocytosis increases—some become large enough to engulf invading protozoa.[65]
In the blood, neutrophils are inactive but are swept along at high speed. When they receive signals from macrophages at the sites of inflammation, they slow down and leave the blood. In the tissues, they are activated by cytokines and arrive at the battle scene ready to kill.[66]
Migration
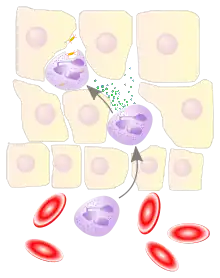
When an infection occurs, a chemical "SOS" signal is given off to attract phagocytes to the site.[67] These chemical signals may include proteins from invading bacteria, clotting system peptides, complement products, and cytokines that have been given off by macrophages located in the tissue near the infection site.[2] Another group of chemical attractants are cytokines that recruit neutrophils and monocytes from the blood.[13]
To reach the site of infection, phagocytes leave the bloodstream and enter the affected tissues. Signals from the infection cause the endothelial cells that line the blood vessels to make a protein called selectin, which neutrophils stick to on passing by. Other signals called vasodilators loosen the junctions connecting endothelial cells, allowing the phagocytes to pass through the wall. Chemotaxis is the process by which phagocytes follow the cytokine "scent" to the infected spot.[2] Neutrophils travel across epithelial cell-lined organs to sites of infection, and although this is an important component of fighting infection, the migration itself can result in disease-like symptoms.[68] During an infection, millions of neutrophils are recruited from the blood, but they die after a few days.[69]
Monocytes
.jpg.webp)
Monocytes develop in the bone marrow and reach maturity in the blood. Mature monocytes have large, smooth, lobed nuclei and abundant cytoplasm that contains granules. Monocytes ingest foreign or dangerous substances and present antigens to other cells of the immune system. Monocytes form two groups: a circulating group and a marginal group that remain in other tissues (approximately 70% are in the marginal group). Most monocytes leave the blood stream after 20–40 hours to travel to tissues and organs and in doing so transform into macrophages[70] or dendritic cells depending on the signals they receive.[71] There are about 500 million monocytes in one litre of human blood.[5]
Macrophages
Mature macrophages do not travel far but stand guard over those areas of the body that are exposed to the outside world. There they act as garbage collectors, antigen presenting cells, or ferocious killers, depending on the signals they receive.[72] They derive from monocytes, granulocyte stem cells, or the cell division of pre-existing macrophages.[73] Human macrophages are about 21 micrometers in diameter.[74]
This type of phagocyte does not have granules but contains many lysosomes. Macrophages are found throughout the body in almost all tissues and organs (e.g., microglial cells in the brain and alveolar macrophages in the lungs), where they silently lie in wait. A macrophage's location can determine its size and appearance. Macrophages cause inflammation through the production of interleukin-1, interleukin-6, and TNF-alpha.[75] Macrophages are usually only found in tissue and are rarely seen in blood circulation. The life-span of tissue macrophages has been estimated to range from four to fifteen days.[76]
Macrophages can be activated to perform functions that a resting monocyte cannot.[75] T helper cells (also known as effector T cells or Th cells), a sub-group of lymphocytes, are responsible for the activation of macrophages. Th1 cells activate macrophages by signaling with IFN-gamma and displaying the protein CD40 ligand.[77] Other signals include TNF-alpha and lipopolysaccharides from bacteria.[75] Th1 cells can recruit other phagocytes to the site of the infection in several ways. They secrete cytokines that act on the bone marrow to stimulate the production of monocytes and neutrophils, and they secrete some of the cytokines that are responsible for the migration of monocytes and neutrophils out of the bloodstream.[78] Th1 cells come from the differentiation of CD4+ T cells once they have responded to antigen in the secondary lymphoid tissues.[75] Activated macrophages play a potent role in tumor destruction by producing TNF-alpha, IFN-gamma, nitric oxide, reactive oxygen compounds, cationic proteins, and hydrolytic enzymes.[75]
Neutrophils
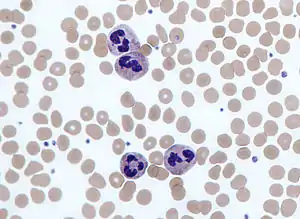
Neutrophils are normally found in the bloodstream and are the most abundant type of phagocyte, constituting 50% to 60% of the total circulating white blood cells.[79] One litre of human blood contains about five billion neutrophils,[5] which are about 10 micrometers in diameter[80] and live for only about five days.[40] Once they have received the appropriate signals, it takes them about thirty minutes to leave the blood and reach the site of an infection.[81] They are ferocious eaters and rapidly engulf invaders coated with antibodies and complement, and damaged cells or cellular debris. Neutrophils do not return to the blood; they turn into pus cells and die.[81] Mature neutrophils are smaller than monocytes and have a segmented nucleus with several sections; each section is connected by chromatin filaments—neutrophils can have 2–5 segments. Neutrophils do not normally exit the bone marrow until maturity but during an infection neutrophil precursors called metamyelocytes, myelocytes and promyelocytes are released.[82]
The intra-cellular granules of the human neutrophil have long been recognized for their protein-destroying and bactericidal properties.[83] Neutrophils can secrete products that stimulate monocytes and macrophages. Neutrophil secretions increase phagocytosis and the formation of reactive oxygen compounds involved in intracellular killing.[84] Secretions from the primary granules of neutrophils stimulate the phagocytosis of IgG-antibody-coated bacteria.[85] When encountering bacteria, fungi or activated platelets they produce web-like chromatin structures known as neutrophil extracellular traps (NETs). Composed mainly of DNA, NETs cause death by a process called netosis – after the pathogens are trapped in NETs they are killed by oxidative and non-oxidative mechanisms.[86]
Dendritic cells
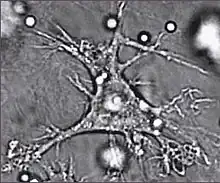
Dendritic cells are specialized antigen-presenting cells that have long outgrowths called dendrites,[87] that help to engulf microbes and other invaders.[88][89] Dendritic cells are present in the tissues that are in contact with the external environment, mainly the skin, the inner lining of the nose, the lungs, the stomach, and the intestines.[90] Once activated, they mature and migrate to the lymphoid tissues where they interact with T cells and B cells to initiate and orchestrate the adaptive immune response.[91] Mature dendritic cells activate T helper cells and cytotoxic T cells.[92] The activated helper T cells interact with macrophages and B cells to activate them in turn. In addition, dendritic cells can influence the type of immune response produced; when they travel to the lymphoid areas where T cells are held they can activate T cells, which then differentiate into cytotoxic T cells or helper T cells.[88]
Mast cells
Mast cells have Toll-like receptors and interact with dendritic cells, B cells, and T cells to help mediate adaptive immune functions.[93] Mast cells express MHC class II molecules and can participate in antigen presentation; however, the mast cell's role in antigen presentation is not very well understood.[94] Mast cells can consume and kill gram-negative bacteria (e.g., salmonella), and process their antigens.[95] They specialize in processing the fimbrial proteins on the surface of bacteria, which are involved in adhesion to tissues.[96][97] In addition to these functions, mast cells produce cytokines that induce an inflammatory response.[98] This is a vital part of the destruction of microbes because the cytokines attract more phagocytes to the site of infection.[95][99]
| Main location | Variety of phenotypes |
|---|---|
| Blood | neutrophils, monocytes |
| Bone marrow | macrophages, monocytes, sinusoidal cells, lining cells |
| Bone tissue | osteoclasts |
| Gut and intestinal Peyer's patches | macrophages |
| Connective tissue | histiocytes, macrophages, monocytes, dendritic cells |
| Liver | Kupffer cells, monocytes |
| Lung | self-replicating macrophages, monocytes, mast cells, dendritic cells |
| Lymphoid tissue | free and fixed macrophages and monocytes, dendritic cells |
| Nervous tissue | microglial cells (CD4+) |
| Spleen | free and fixed macrophages, monocytes, sinusoidal cells |
| Thymus | free and fixed macrophages and monocytes |
| Skin | resident Langerhans cells, other dendritic cells, conventional macrophages, mast cells |
Non-professional phagocytes
Dying cells and foreign organisms are consumed by cells other than the "professional" phagocytes.[101] These cells include epithelial cells, endothelial cells, fibroblasts, and mesenchymal cells. They are called non-professional phagocytes, to emphasize that, in contrast to professional phagocytes, phagocytosis is not their principal function.[102] Fibroblasts, for example, which can phagocytose collagen in the process of remolding scars, will also make some attempt to ingest foreign particles.[103]
Non-professional phagocytes are more limited than professional phagocytes in the type of particles they can take up. This is due to their lack of efficient phagocytic receptors, in particular opsonins—which are antibodies and complement attached to invaders by the immune system.[11] Additionally, most non-professional phagocytes do not produce reactive oxygen-containing molecules in response to phagocytosis.[104]
| Main location | Variety of phenotypes |
|---|---|
| Blood, lymph and lymph nodes | Lymphocytes |
| Blood, lymph and lymph nodes | NK and LGL cells (large granular lymphocytes) |
| Blood | Eosinophils and Basophils[105] |
| Skin | Epithelial cells |
| Liver | Hepatocytes[106] |
| Blood vessels | Endothelial cells |
| Connective tissue | Fibroblasts |
Pathogen evasion and resistance

A pathogen is only successful in infecting an organism if it can get past its defenses. Pathogenic bacteria and protozoa have developed a variety of methods to resist attacks by phagocytes, and many actually survive and replicate within phagocytic cells.[107][108]
Avoiding contact
There are several ways bacteria avoid contact with phagocytes. First, they can grow in sites that phagocytes are not capable of traveling to (e.g., the surface of unbroken skin). Second, bacteria can suppress the inflammatory response; without this response to infection phagocytes cannot respond adequately. Third, some species of bacteria can inhibit the ability of phagocytes to travel to the site of infection by interfering with chemotaxis.[107] Fourth, some bacteria can avoid contact with phagocytes by tricking the immune system into "thinking" that the bacteria are "self". Treponema pallidum—the bacterium that causes syphilis—hides from phagocytes by coating its surface with fibronectin,[109] which is produced naturally by the body and plays a crucial role in wound healing.[110]
Avoiding engulfment
Bacteria often produce capsules made of proteins or sugars that coat their cells and interfere with phagocytosis.[107] Some examples are the K5 capsule and O75 O antigen found on the surface of Escherichia coli,[111] and the exopolysaccharide capsules of Staphylococcus epidermidis.[112] Streptococcus pneumoniae produces several types of capsule that provide different levels of protection,[113] and group A streptococci produce proteins such as M protein and fimbrial proteins to block engulfment. Some proteins hinder opsonin-related ingestion; Staphylococcus aureus produces Protein A to block antibody receptors, which decreases the effectiveness of opsonins.[114] Enteropathogenic species of the genus Yersinia bind with the use of the virulence factor YopH to receptors of phagocytes from which they influence the cells capability to exert phagocytosis.[115]
Survival inside the phagocyte
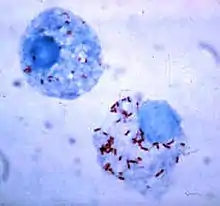
Bacteria have developed ways to survive inside phagocytes, where they continue to evade the immune system.[116] To get safely inside the phagocyte they express proteins called invasins. When inside the cell they remain in the cytoplasm and avoid toxic chemicals contained in the phagolysosomes.[117] Some bacteria prevent the fusion of a phagosome and lysosome, to form the phagolysosome.[107] Other pathogens, such as Leishmania, create a highly modified vacuole inside the phagocyte, which helps them persist and replicate.[118] Some bacteria are capable of living inside of the phagolysosome. Staphylococcus aureus, for example, produces the enzymes catalase and superoxide dismutase, which break down chemicals—such as hydrogen peroxide—produced by phagocytes to kill bacteria.[119] Bacteria may escape from the phagosome before the formation of the phagolysosome: Listeria monocytogenes can make a hole in the phagosome wall using enzymes called listeriolysin O and phospholipase C.[120]
Killing
Bacteria have developed several ways of killing phagocytes.[114] These include cytolysins, which form pores in the phagocyte's cell membranes, streptolysins and leukocidins, which cause neutrophils' granules to rupture and release toxic substances,[121][122] and exotoxins that reduce the supply of a phagocyte's ATP, needed for phagocytosis. After a bacterium is ingested, it may kill the phagocyte by releasing toxins that travel through the phagosome or phagolysosome membrane to target other parts of the cell.[107]
Disruption of cell signaling
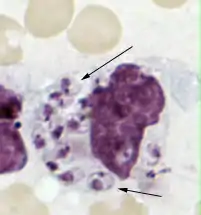
Some survival strategies often involve disrupting cytokines and other methods of cell signaling to prevent the phagocyte's responding to invasion.[123] The protozoan parasites Toxoplasma gondii, Trypanosoma cruzi, and Leishmania infect macrophages, and each has a unique way of taming them.[123] Some species of Leishmania alter the infected macrophage's signalling, repress the production of cytokines and microbicidal molecules—nitric oxide and reactive oxygen species—and compromise antigen presentation.[124]
Host damage by phagocytes
Macrophages and neutrophils, in particular, play a central role in the inflammatory process by releasing proteins and small-molecule inflammatory mediators that control infection but can damage host tissue. In general, phagocytes aim to destroy pathogens by engulfing them and subjecting them to a battery of toxic chemicals inside a phagolysosome. If a phagocyte fails to engulf its target, these toxic agents can be released into the environment (an action referred to as "frustrated phagocytosis"). As these agents are also toxic to host cells, they can cause extensive damage to healthy cells and tissues.[125]
When neutrophils release their granule contents in the kidney, the contents of the granule (reactive oxygen compounds and proteases) degrade the extracellular matrix of host cells and can cause damage to glomerular cells, affecting their ability to filter blood and causing changes in shape. In addition, phospholipase products (e.g., leukotrienes) intensify the damage. This release of substances promotes chemotaxis of more neutrophils to the site of infection, and glomerular cells can be damaged further by the adhesion molecules during the migration of neutrophils. The injury done to the glomerular cells can cause kidney failure.[126]
Neutrophils also play a key role in the development of most forms of acute lung injury.[127] Here, activated neutrophils release the contents of their toxic granules into the lung environment.[128] Experiments have shown that a reduction in the number of neutrophils lessens the effects of acute lung injury,[129] but treatment by inhibiting neutrophils is not clinically realistic, as it would leave the host vulnerable to infection.[128] In the liver, damage by neutrophils can contribute to dysfunction and injury in response to the release of endotoxins produced by bacteria, sepsis, trauma, alcoholic hepatitis, ischemia, and hypovolemic shock resulting from acute hemorrhage.[130]
Chemicals released by macrophages can also damage host tissue. TNF-α is an important chemical that is released by macrophages that causes the blood in small vessels to clot to prevent an infection from spreading.[131] If a bacterial infection spreads to the blood, TNF-α is released into vital organs, which can cause vasodilation and a decrease in plasma volume; these in turn can be followed by septic shock. During septic shock, TNF-α release causes a blockage of the small vessels that supply blood to the vital organs, and the organs may fail. Septic shock can lead to death.[13]
Evolutionary origins
_(52606801786).jpg.webp)
Phagocytosis is common and probably appeared early in evolution,[132] evolving first in unicellular eukaryotes.[133] Amoebae are unicellular protists that separated from the tree leading to metazoa shortly after the divergence of plants, and they share many specific functions with mammalian phagocytic cells.[133] Dictyostelium discoideum, for example, is an amoeba that lives in the soil and feeds on bacteria. Like animal phagocytes, it engulfs bacteria by phagocytosis mainly through Toll-like receptors, and it has other biological functions in common with macrophages.[134] Dictyostelium discoideum is social; it aggregates when starved to form a migrating pseudoplasmodium or slug. This multicellular organism eventually will produce a fruiting body with spores that are resistant to environmental dangers. Before the formation of fruiting bodies, the cells will migrate as a slug-like organism for several days. During this time, exposure to toxins or bacterial pathogens has the potential to compromise survival of the species by limiting spore production. Some of the amoebae engulf bacteria and absorb toxins while circulating within the slug, and these amoebae eventually die. They are genetically identical to the other amoebae in the slug; their self-sacrifice to protect the other amoebae from bacteria is similar to the self-sacrifice of phagocytes seen in the immune system of higher vertebrates. This ancient immune function in social amoebae suggests an evolutionarily conserved cellular foraging mechanism that might have been adapted to defense functions well before the diversification of amoebae into higher forms.[135] Phagocytes occur throughout the animal kingdom,[3] from marine sponges to insects and lower and higher vertebrates.[136][137] The ability of amoebae to distinguish between self and non-self is a pivotal one, and is the root of the immune system of many species of amoeba.[8]
References
- Little C, Fowler HW, Coulson J (1983). The Shorter Oxford English Dictionary. Oxford University Press (Guild Publishing). pp. 1566–67.
- Delves et al. 2006, pp. 2–10
- Delves et al. 2006, p. 250
- Delves et al. 2006, p. 251
- Hoffbrand, Pettit & Moss 2005, p. 331
- Ilya Mechnikov, retrieved on November 28, 2008. From Nobel Lectures, Physiology or Medicine 1901–1921, Elsevier Publishing Company, Amsterdam, 1967. Archived August 22, 2008, at the Wayback Machine
- Schmalstieg, FC; AS Goldman (2008). "Ilya Ilich Metchnikoff (1845–1915) and Paul Ehrlich (1854–1915): the centennial of the 1908 Nobel Prize in Physiology or Medicine". Journal of Medical Biography. 16 (2): 96–103. doi:10.1258/jmb.2008.008006. PMID 18463079. S2CID 25063709.
- Janeway, Chapter: Evolution of the innate immune system. retrieved on March 20, 2009
- Ernst & Stendahl 2006, p. 186
- Robinson & Babcock 1998, p. 187 and Ernst & Stendahl 2006, pp. 7–10
- Ernst & Stendahl 2006, p. 10
- Thompson CB (1995). "Apoptosis in the pathogenesis and treatment of disease". Science. 267 (5203): 1456–62. Bibcode:1995Sci...267.1456T. doi:10.1126/science.7878464. PMID 7878464. S2CID 12991980.
- Janeway, Chapter: Induced innate responses to infection.
- Fang FC (October 2004). "Antimicrobial reactive oxygen and nitrogen species: concepts and controversies". Nat. Rev. Microbiol. 2 (10): 820–32. doi:10.1038/nrmicro1004. PMID 15378046. S2CID 11063073.
- Delves et al. 2006, pp. 172–84
- Kaufmann SH (2019). "Immunology's Coming of Age". Frontiers in Immunology. 10: 684. doi:10.3389/fimmu.2019.00684. PMC 6456699. PMID 31001278.
- Aterman K (April 1, 1998). "Medals, memoirs—and Metchnikoff". J. Leukoc. Biol. 63 (4): 515–17. doi:10.1002/jlb.63.4.515. PMID 9544583. S2CID 44748502.
- "Ilya Mechnikov". The Nobel Foundation. Retrieved December 19, 2014.
- Delves et al. 2006, p. 263
- Robinson & Babcock 1998, p. vii
- Ernst & Stendahl 2006, p. 6
- Ernst & Stendahl 2006, p. 4
- Ernst & Stendahl 2006, p. 78
- Feldman MB, Vyas JM, Mansour MK (May 2019). "It takes a village: Phagocytes play a central role in fungal immunity". Seminars in Cell & Developmental Biology. 89: 16–23. doi:10.1016/j.semcdb.2018.04.008. PMC 6235731. PMID 29727727.
- Hampton MB, Vissers MC, Winterbourn CC (February 1994). "A single assay for measuring the rates of phagocytosis and bacterial killing by neutrophils". J. Leukoc. Biol. 55 (2): 147–52. doi:10.1002/jlb.55.2.147. PMID 8301210. S2CID 44911791. Archived from the original on December 28, 2012. Retrieved December 19, 2014.
- Delves et al. 2006, pp. 6–7
- Sompayrac 2019, p. 2
- Sompayrac 2019, p. 2
- Sompayrac 2019, pp. 13–16
- Freund I, Eigenbrod T, Helm M, Dalpke AH (January 2019). "RNA Modifications Modulate Activation of Innate Toll-Like Receptors". Genes. 10 (2): 92. doi:10.3390/genes10020092. PMC 6410116. PMID 30699960.
- Dale DC, Boxer L, Liles WC (August 2008). "The phagocytes: neutrophils and monocytes". Blood. 112 (4): 935–45. doi:10.1182/blood-2007-12-077917. PMID 18684880. S2CID 746699.
- Dahlgren, C; A Karlsson (December 17, 1999). "Respiratory burst in human neutrophils". Journal of Immunological Methods. 232 (1–2): 3–14. doi:10.1016/S0022-1759(99)00146-5. PMID 10618505.
- Shatwell, KP; AW Segal (1996). "NADPH oxidase". The International Journal of Biochemistry & Cell Biology. 28 (11): 1191–95. doi:10.1016/S1357-2725(96)00084-2. PMID 9022278.
- Klebanoff SJ (1999). "Myeloperoxidase". Proc. Assoc. Am. Physicians. 111 (5): 383–89. doi:10.1111/paa.1999.111.5.383. PMID 10519157.
- Meyer KC (September 2004). "Neutrophils, myeloperoxidase, and bronchiectasis in cystic fibrosis: green is not good". J. Lab. Clin. Med. 144 (3): 124–26. doi:10.1016/j.lab.2004.05.014. PMID 15478278.
- Hoffbrand, Pettit & Moss 2005, p. 118
- Delves et al. 2006, pp. 6–10
- Schroder K, Hertzog PJ, Ravasi T, Hume DA (February 2004). "Interferon-gamma: an overview of signals, mechanisms and functions". J. Leukoc. Biol. 75 (2): 163–89. doi:10.1189/jlb.0603252. PMID 14525967. S2CID 15862242.
- Delves et al. 2006, p. 188
- Sompayrac 2019, p. 136
- Lipu HN, Ahmed TA, Ali S, Ahmed D, Waqar MA (September 2008). "Chronic granulomatous disease". J Pak Med Assoc. 58 (9): 516–18. PMID 18846805.
- Kaplan J, De Domenico I, Ward DM (January 2008). "Chediak-Higashi syndrome". Curr. Opin. Hematol. 15 (1): 22–29. doi:10.1097/MOH.0b013e3282f2bcce. PMID 18043242. S2CID 43243529.
- Sompayrac 2019, p. 7
- de Almeida SM, Nogueira MB, Raboni SM, Vidal LR (October 2007). "Laboratorial diagnosis of lymphocytic meningitis". Braz J Infect Dis. 11 (5): 489–95. doi:10.1590/s1413-86702007000500010. PMID 17962876.
- Sompayrac 2019, p. 22
- Sompayrac 2019, p. 68
- "Apoptosis". Merriam-Webster Online Dictionary. Retrieved December 19, 2014.
- Li MO, Sarkisian MR, Mehal WZ, Rakic P, Flavell RA (November 2003). "Phosphatidylserine receptor is required for clearance of apoptotic cells". Science. 302 (5650): 1560–63. doi:10.1126/science.1087621. PMID 14645847. S2CID 36252352. (Free registration required for online access)
- Nagata S, Sakuragi T, Segawa K (December 2019). "Flippase and scramblase for phosphatidylserine exposure". Current Opinion in Immunology. 62: 31–38. doi:10.1016/j.coi.2019.11.009. PMID 31837595.
- Wang X (2003). "Cell corpse engulfment mediated by C. elegans phosphatidylserine receptor through CED-5 and CED-12". Science. 302 (5650): 1563–1566. Bibcode:2003Sci...302.1563W. doi:10.1126/science.1087641. PMID 14645848. S2CID 25672278. (Free registration required for online access)
- Savill J, Gregory C, Haslett C (2003). "Eat me or die". Science. 302 (5650): 1516–17. doi:10.1126/science.1092533. hdl:1842/448. PMID 14645835. S2CID 13402617.
- Zhou Z, Yu X (October 2008). "Phagosome maturation during the removal of apoptotic cells: receptors lead the way". Trends Cell Biol. 18 (10): 474–85. doi:10.1016/j.tcb.2008.08.002. PMC 3125982. PMID 18774293.
- Sompayrac 2019, p. 3
- Sompayrac 2019, p. 4
- Sompayrac 2019, pp. 27–35
- Delves et al. 2006, pp. 171–184
- Delves et al. 2006, pp. 456
- Lee T, McGibbon A (2004). "Antigen Presenting Cells (APC)". Dalhousie University. Archived from the original on January 12, 2008. Retrieved December 19, 2014.
- Delves et al. 2006, p. 161
- Sompayrac 2019, p. 8
- Delves et al. 2006, pp. 237–242
- Lange C, Dürr M, Doster H, Melms A, Bischof F (2007). "Dendritic cell-regulatory T-cell interactions control self-directed immunity". Immunol. Cell Biol. 85 (8): 575–81. doi:10.1038/sj.icb.7100088. PMID 17592494. S2CID 36342899.
- Steinman, Ralph M. (2004). "Dendritic Cells and Immune Tolerance". The Rockefeller University. Archived from the original on March 11, 2009. Retrieved December 19, 2014.
- Romagnani, S (2006). "Immunological tolerance and autoimmunity". Internal and Emergency Medicine. 1 (3): 187–96. doi:10.1007/BF02934736. PMID 17120464. S2CID 27585046.
- Sompayrac 2019, pp. 16–17
- Sompayrac 2019, pp. 18–19
- Delves et al. 2006, p. 6
- Zen K, Parkos CA (October 2003). "Leukocyte-epithelial interactions". Curr. Opin. Cell Biol. 15 (5): 557–64. doi:10.1016/S0955-0674(03)00103-0. PMID 14519390.
- Sompayrac 2019, p. 18
- Hoffbrand, Pettit & Moss 2005, p. 117
- Delves et al. 2006, pp. 1–6
- Sompayrac 2019, p. 136
- Takahashi K, Naito M, Takeya M (July 1996). "Development and heterogeneity of macrophages and their related cells through their differentiation pathways". Pathol. Int. 46 (7): 473–85. doi:10.1111/j.1440-1827.1996.tb03641.x. PMID 8870002. S2CID 6049656.
- Krombach F, Münzing S, Allmeling AM, Gerlach JT, Behr J, Dörger M (September 1997). "Cell size of alveolar macrophages: an interspecies comparison". Environ. Health Perspect. 105 (Suppl 5): 1261–63. doi:10.2307/3433544. JSTOR 3433544. PMC 1470168. PMID 9400735.
- Delves et al. 2006, pp. 31–36
- Ernst & Stendahl 2006, p. 8
- Delves et al. 2006, p. 156
- Delves et al. 2006, p. 187
- Stvrtinová, Viera; Ján Jakubovský and Ivan Hulín (1995). "Neutrophils, central cells in acute inflammation". Inflammation and Fever from Pathophysiology: Principles of Disease. Computing Centre, Slovak Academy of Sciences: Academic Electronic Press. ISBN 978-80-967366-1-4. Archived from the original on December 31, 2010. Retrieved December 19, 2014.
- Delves et al. 2006, p. 4
- Sompayrac 2019, p. 18
- Linderkamp O, Ruef P, Brenner B, Gulbins E, Lang F (December 1998). "Passive deformability of mature, immature, and active neutrophils in healthy and septicemic neonates". Pediatr. Res. 44 (6): 946–50. doi:10.1203/00006450-199812000-00021. PMID 9853933.
- Paoletti, Notario & Ricevuti 1997, p. 62
- Soehnlein O, Kenne E, Rotzius P, Eriksson EE, Lindbom L (January 2008). "Neutrophil secretion products regulate anti-bacterial activity in monocytes and macrophages". Clin. Exp. Immunol. 151 (1): 139–45. doi:10.1111/j.1365-2249.2007.03532.x. PMC 2276935. PMID 17991288.
- Soehnlein O, Kai-Larsen Y, Frithiof R (October 2008). "Neutrophil primary granule proteins HBP and HNP1-3 boost bacterial phagocytosis by human and murine macrophages". J. Clin. Invest. 118 (10): 3491–502. doi:10.1172/JCI35740. PMC 2532980. PMID 18787642.
- Papayannopoulos V (February 2018). "Neutrophil extracellular traps in immunity and disease". Nature Reviews. Immunology. 18 (2): 134–147. doi:10.1038/nri.2017.105. PMID 28990587. S2CID 25067858.
- Steinman RM, Cohn ZA (1973). "Identification of a novel cell type in peripheral lymphoid organs of mice. I. Morphology, quantitation, tissue distribution". J. Exp. Med. 137 (5): 1142–62. doi:10.1084/jem.137.5.1142. PMC 2139237. PMID 4573839.
- Steinman, Ralph. "Dendritic Cells". The Rockefeller University. Archived from the original on June 27, 2009. Retrieved December 19, 2014.
- Guermonprez P, Valladeau J, Zitvogel L, Théry C, Amigorena S (2002). "Antigen presentation and T cell stimulation by dendritic cells". Annu. Rev. Immunol. 20: 621–67. doi:10.1146/annurev.immunol.20.100301.064828. PMID 11861614.
- Hoffbrand, Pettit & Moss 2005, p. 134
- Sallusto F, Lanzavecchia A (2002). "The instructive role of dendritic cells on T-cell responses". Arthritis Res. 4 (Suppl 3): S127–32. doi:10.1186/ar567. PMC 3240143. PMID 12110131.
- Sompayrac 2019, pp. 45–46
- Novak N, Bieber T, Peng WM (2010). "The immunoglobulin E-Toll-like receptor network". International Archives of Allergy and Immunology. 151 (1): 1–7. doi:10.1159/000232565. PMID 19672091. Retrieved December 19, 2014.
- Kalesnikoff J, Galli SJ (November 2008). "New developments in mast cell biology". Nature Immunology. 9 (11): 1215–23. doi:10.1038/ni.f.216. PMC 2856637. PMID 18936782.
- Malaviya R, Abraham SN (February 2001). "Mast cell modulation of immune responses to bacteria". Immunol. Rev. 179: 16–24. doi:10.1034/j.1600-065X.2001.790102.x. PMID 11292019. S2CID 23115222.
- Connell I, Agace W, Klemm P, Schembri M, Mărild S, Svanborg C (September 1996). "Type 1 fimbrial expression enhances Escherichia coli virulence for the urinary tract". Proc. Natl. Acad. Sci. U.S.A. 93 (18): 9827–32. Bibcode:1996PNAS...93.9827C. doi:10.1073/pnas.93.18.9827. PMC 38514. PMID 8790416.
- Malaviya R, Twesten NJ, Ross EA, Abraham SN, Pfeifer JD (February 1996). "Mast cells process bacterial Ags through a phagocytic route for class I MHC presentation to T cells". J. Immunol. 156 (4): 1490–96. doi:10.4049/jimmunol.156.4.1490. PMID 8568252. S2CID 7917861. Retrieved December 19, 2014.
- Taylor ML, Metcalfe DD (2001). "Mast cells in allergy and host defense". Allergy Asthma Proc. 22 (3): 115–19. doi:10.2500/108854101778148764. PMID 11424870.
- Urb M, Sheppard DC (2012). "The role of mast cells in the defence against pathogens". PLOS Pathogens. 8 (4): e1002619. doi:10.1371/journal.ppat.1002619. PMC 3343118. PMID 22577358.
- Paoletti, Notario & Ricevuti 1997, p. 427
- Birge RB, Ucker DS (July 2008). "Innate apoptotic immunity: the calming touch of death". Cell Death Differ. 15 (7): 1096–1102. doi:10.1038/cdd.2008.58. PMID 18451871.
- Couzinet S, Cejas E, Schittny J, Deplazes P, Weber R, Zimmerli S (December 2000). "Phagocytic uptake of Encephalitozoon cuniculi by nonprofessional phagocytes". Infect. Immun. 68 (12): 6939–45. doi:10.1128/IAI.68.12.6939-6945.2000. PMC 97802. PMID 11083817.
- Segal G, Lee W, Arora PD, McKee M, Downey G, McCulloch CA (January 2001). "Involvement of actin filaments and integrins in the binding step in collagen phagocytosis by human fibroblasts". Journal of Cell Science. 114 (Pt 1): 119–129. doi:10.1242/jcs.114.1.119. PMID 11112696.
- Rabinovitch M (March 1995). "Professional and non-professional phagocytes: an introduction". Trends Cell Biol. 5 (3): 85–87. doi:10.1016/S0962-8924(00)88955-2. PMID 14732160.
- Lin A, Loré K (2017). "Granulocytes: New Members of the Antigen-Presenting Cell Family". Frontiers in Immunology. 8: 1781. doi:10.3389/fimmu.2017.01781. PMC 5732227. PMID 29321780.
- Davies SP, Terry LV, Wilkinson AL, Stamataki Z (2020). "Cell-in-Cell Structures in the Liver: A Tale of Four E's". Frontiers in Immunology. 11: 650. doi:10.3389/fimmu.2020.00650. PMC 7247839. PMID 32528462.
- Todar, Kenneth. "Mechanisms of Bacterial Pathogenicity: Bacterial Defense Against Phagocytes". 2008. Retrieved December 19, 2014.
- Alexander J, Satoskar AR, Russell DG (September 1999). "Leishmania species: models of intracellular parasitism". J. Cell Sci. 112 (18): 2993–3002. doi:10.1242/jcs.112.18.2993. PMID 10462516. Retrieved December 19, 2014.
- Celli J, Finlay BB (May 2002). "Bacterial avoidance of phagocytosis". Trends Microbiol. 10 (5): 232–37. doi:10.1016/S0966-842X(02)02343-0. PMID 11973157.
- Valenick LV, Hsia HC, Schwarzbauer JE (September 2005). "Fibronectin fragmentation promotes alpha4beta1 integrin-mediated contraction of a fibrin-fibronectin provisional matrix". Experimental Cell Research. 309 (1): 48–55. doi:10.1016/j.yexcr.2005.05.024. PMID 15992798.
- Burns SM, Hull SI (August 1999). "Loss of resistance to ingestion and phagocytic killing by O(-) and K(-) mutants of a uropathogenic Escherichia coli O75:K5 strain". Infect. Immun. 67 (8): 3757–62. doi:10.1128/IAI.67.8.3757-3762.1999. PMC 96650. PMID 10417134.
- Vuong C, Kocianova S, Voyich JM (December 2004). "A crucial role for exopolysaccharide modification in bacterial biofilm formation, immune evasion, and virulence". J. Biol. Chem. 279 (52): 54881–86. doi:10.1074/jbc.M411374200. PMID 15501828.
- Melin M, Jarva H, Siira L, Meri S, Käyhty H, Väkeväinen M (February 2009). "Streptococcus pneumoniae capsular serotype 19F is more resistant to C3 deposition and less sensitive to opsonophagocytosis than serotype 6B". Infect. Immun. 77 (2): 676–84. doi:10.1128/IAI.01186-08. PMC 2632042. PMID 19047408.
- Foster TJ (December 2005). "Immune evasion by staphylococci". Nat. Rev. Microbiol. 3 (12): 948–58. doi:10.1038/nrmicro1289. PMID 16322743. S2CID 205496221.
- Fällman M, Deleuil F, McGee K (February 2002). "Resistance to phagocytosis by Yersinia". International Journal of Medical Microbiology. 291 (6–7): 501–9. doi:10.1078/1438-4221-00159. PMID 11890550.
- Sansonetti P (December 2001). "Phagocytosis of bacterial pathogens: implications in the host response". Semin. Immunol. 13 (6): 381–90. doi:10.1006/smim.2001.0335. PMID 11708894.
- Dersch P, Isberg RR (March 1999). "A region of the Yersinia pseudotuberculosis invasin protein enhances integrin-mediated uptake into mammalian cells and promotes self-association". EMBO J. 18 (5): 1199–1213. doi:10.1093/emboj/18.5.1199. PMC 1171211. PMID 10064587.
- Antoine JC, Prina E, Lang T, Courret N (October 1998). "The biogenesis and properties of the parasitophorous vacuoles that harbour Leishmania in murine macrophages". Trends Microbiol. 6 (10): 392–401. doi:10.1016/S0966-842X(98)01324-9. PMID 9807783.
- Das D, Saha SS, Bishayi B (July 2008). "Intracellular survival of Staphylococcus aureus: correlating production of catalase and superoxide dismutase with levels of inflammatory cytokines". Inflamm. Res. 57 (7): 340–49. doi:10.1007/s00011-007-7206-z. PMID 18607538. S2CID 22127111.
- Hara H, Kawamura I, Nomura T, Tominaga T, Tsuchiya K, Mitsuyama M (August 2007). "Cytolysin-dependent escape of the bacterium from the phagosome is required but not sufficient for induction of the Th1 immune response against Listeria monocytogenes infection: distinct role of Listeriolysin O determined by cytolysin gene replacement". Infect. Immun. 75 (8): 3791–3801. doi:10.1128/IAI.01779-06. PMC 1951982. PMID 17517863.
- Datta V, Myskowski SM, Kwinn LA, Chiem DN, Varki N, Kansal RG, Kotb M, Nizet V (May 2005). "Mutational analysis of the group A streptococcal operon encoding streptolysin S and its virulence role in invasive infection". Mol. Microbiol. 56 (3): 681–95. doi:10.1111/j.1365-2958.2005.04583.x. PMID 15819624. S2CID 14748436.
- Iwatsuki K, Yamasaki O, Morizane S, Oono T (June 2006). "Staphylococcal cutaneous infections: invasion, evasion and aggression". J. Dermatol. Sci. 42 (3): 203–14. doi:10.1016/j.jdermsci.2006.03.011. PMID 16679003.
- Denkers EY, Butcher BA (January 2005). "Sabotage and exploitation in macrophages parasitized by intracellular protozoans". Trends Parasitol. 21 (1): 35–41. doi:10.1016/j.pt.2004.10.004. PMID 15639739.
- Gregory DJ, Olivier M (2005). "Subversion of host cell signalling by the protozoan parasite Leishmania". Parasitology. 130 Suppl: S27–35. doi:10.1017/S0031182005008139. PMID 16281989. S2CID 24696519.
- Paoletti pp. 426–30
- Heinzelmann M, Mercer-Jones MA, Passmore JC (August 1999). "Neutrophils and renal failure". Am. J. Kidney Dis. 34 (2): 384–99. doi:10.1016/S0272-6386(99)70375-6. PMID 10430993.
- Lee WL, Downey GP (February 2001). "Neutrophil activation and acute lung injury". Curr Opin Crit Care. 7 (1): 1–7. doi:10.1097/00075198-200102000-00001. PMID 11373504. S2CID 24164360.
- Moraes TJ, Zurawska JH, Downey GP (January 2006). "Neutrophil granule contents in the pathogenesis of lung injury". Curr. Opin. Hematol. 13 (1): 21–27. doi:10.1097/01.moh.0000190113.31027.d5. PMID 16319683. S2CID 29374195.
- Abraham E (April 2003). "Neutrophils and acute lung injury". Crit. Care Med. 31 (4 Suppl): S195–99. doi:10.1097/01.CCM.0000057843.47705.E8. PMID 12682440. S2CID 4004607.
- Ricevuti G (December 1997). "Host tissue damage by phagocytes". Ann. N. Y. Acad. Sci. 832 (1): 426–48. Bibcode:1997NYASA.832..426R. doi:10.1111/j.1749-6632.1997.tb46269.x. PMID 9704069. S2CID 10318084.
- Charley B, Riffault S, Van Reeth K (October 2006). "Porcine innate and adaptative immune responses to influenza and coronavirus infections". Ann. N. Y. Acad. Sci. 1081 (1): 130–36. Bibcode:2006NYASA1081..130C. doi:10.1196/annals.1373.014. hdl:1854/LU-369324. PMC 7168046. PMID 17135502.
- Sompayrac 2019, p. 2
- Cosson P, Soldati T (June 2008). "Eat, kill or die: when amoeba meets bacteria". Curr. Opin. Microbiol. 11 (3): 271–76. doi:10.1016/j.mib.2008.05.005. PMID 18550419.
- Bozzaro S, Bucci C, Steinert M (2008). "Phagocytosis and host-pathogen interactions in Dictyostelium with a look at macrophages". International Review of Cell and Molecular Biology. Vol. 271. pp. 253–300. doi:10.1016/S1937-6448(08)01206-9. ISBN 978-0-12-374728-0. PMID 19081545. S2CID 7326149.
- Chen G, Zhuchenko O, Kuspa A (August 2007). "Immune-like phagocyte activity in the social amoeba". Science. 317 (5838): 678–81. Bibcode:2007Sci...317..678C. doi:10.1126/science.1143991. PMC 3291017. PMID 17673666.
- Delves et al. 2006, pp. 251–252
- Hanington PC, Tam J, Katzenback BA, Hitchen SJ, Barreda DR, Belosevic M (April 2009). "Development of macrophages of cyprinid fish". Dev. Comp. Immunol. 33 (4): 411–29. doi:10.1016/j.dci.2008.11.004. PMID 19063916.
Bibliography
- Delves, P. J.; Martin, S. J.; Burton, D. R.; Roit, I. M. (2006). Roitt's Essential Immunology (11th ed.). Malden, MA: Blackwell Publishing. ISBN 978-1-4051-3603-7.
- Ernst, J. D.; Stendahl, O., eds. (2006). Phagocytosis of Bacteria and Bacterial Pathogenicity. New York: Cambridge University Press. ISBN 978-0-521-84569-4. Website
- Hoffbrand, A. V.; Pettit, J. E.; Moss, P. A. H. (2005). Essential Haematology (4th ed.). London: Blackwell Science. ISBN 978-0-632-05153-3.
- Paoletti, R.; Notario, A.; Ricevuti, G., eds. (1997). Phagocytes: Biology, Physiology, Pathology, and Pharmacotherapeutics. New York: The New York Academy of Sciences. ISBN 978-1-57331-102-1.
- Robinson, J. P.; Babcock, G. F., eds. (1998). Phagocyte Function — A guide for research and clinical evaluation. New York: Wiley–Liss. ISBN 978-0-471-12364-4.
- Sompayrac, L. (2019). How the Immune System Works (6th ed.). Malden, MA: Blackwell Publishing. ISBN 978-1-119-54212-4.
External links
| Library resources about Phagocyte |
- Phagocytes at the U.S. National Library of Medicine Medical Subject Headings (MeSH)
- White blood cell engulfing bacteria