Photochemistry
Photochemistry is the branch of chemistry concerned with the chemical effects of light. Generally, this term is used to describe a chemical reaction caused by absorption of ultraviolet (wavelength from 100 to 400 nm), visible light (400–750 nm) or infrared radiation (750–2500 nm).[1]

In nature, photochemistry is of immense importance as it is the basis of photosynthesis, vision, and the formation of vitamin D with sunlight.[2] It is also responsible for the appearance of DNA mutations leading to skin cancers.[3]
Photochemical reactions proceed differently than temperature-driven reactions. Photochemical paths access high energy intermediates that cannot be generated thermally, thereby overcoming large activation barriers in a short period of time, and allowing reactions otherwise inaccessible by thermal processes. Photochemistry can also be destructive, as illustrated by the photodegradation of plastics.
Concept
Grotthuss–Draper law and Stark–Einstein law
Photoexcitation is the first step in a photochemical process where the reactant is elevated to a state of higher energy, an excited state. The first law of photochemistry, known as the Grotthuss–Draper law (for chemists Theodor Grotthuss and John W. Draper), states that light must be absorbed by a chemical substance in order for a photochemical reaction to take place. According to the second law of photochemistry, known as the Stark–Einstein law (for physicists Johannes Stark and Albert Einstein), for each photon of light absorbed by a chemical system, no more than one molecule is activated for a photochemical reaction, as defined by the quantum yield.[4][5]
Fluorescence and phosphorescence
When a molecule or atom in the ground state (S0) absorbs light, one electron is excited to a higher orbital level. This electron maintains its spin according to the spin selection rule; other transitions would violate the law of conservation of angular momentum. The excitation to a higher singlet state can be from HOMO to LUMO or to a higher orbital, so that singlet excitation states S1, S2, S3... at different energies are possible.
Kasha's rule stipulates that higher singlet states would quickly relax by radiationless decay or internal conversion (IC) to S1. Thus, S1 is usually, but not always, the only relevant singlet excited state. This excited state S1 can further relax to S0 by IC, but also by an allowed radiative transition from S1 to S0 that emits a photon; this process is called fluorescence.
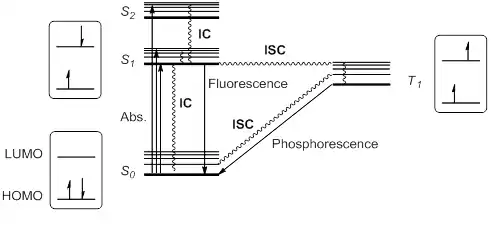
Alternatively, it is possible for the excited state S1 to undergo spin inversion and to generate a triplet excited state T1 having two unpaired electrons with the same spin. This violation of the spin selection rule is possible by intersystem crossing (ISC) of the vibrational and electronic levels of S1 and T1. According to Hund's rule of maximum multiplicity, this T1 state would be somewhat more stable than S1.
This triplet state can relax to the ground state S0 by radiationless IC or by a radiation pathway called phosphorescence. This process implies a change of electronic spin, which is forbidden by spin selection rules, making phosphorescence (from T1 to S0) much slower than fluorescence (from S1 to S0). Thus, triplet states generally have longer lifetimes than singlet states. These transitions are usually summarized in a state energy diagram or Jablonski diagram, the paradigm of molecular photochemistry.
These excited species, either S1 or T1, have a half empty low-energy orbital, and are consequently more oxidizing than the ground state. But at the same time, they have an electron in a high energy orbital, and are thus more reducing. In general, excited species are prone to participate in electron transfer processes.[6]
Experimental set-up

Photochemical reactions require a light source that emits wavelengths corresponding to an electronic transition in the reactant. In the early experiments (and in everyday life), sunlight was the light source, although it is polychromatic. Mercury-vapor lamps are more common in the laboratory. Low pressure mercury vapor lamps mainly emit at 254 nm. For polychromatic sources, wavelength ranges can be selected using filters. Alternatively, laser beams are usually monochromatic (although two or more wavelengths can be obtained using nonlinear optics) and LEDs have a relatively narrowband that can be efficiently used, as well as Rayonet lamps, to get approximately monochromatic beams.
9SchlenkCropped.png.webp)
The emitted light must of course reach the targeted functional group without being blocked by the reactor, medium, or other functional groups present. For many applications, quartz is used for the reactors as well as to contain the lamp. Pyrex absorbs at wavelengths shorter than 275 nm. The solvent is an important experimental parameter. Solvents are potential reactants and for this reason, chlorinated solvents are avoided because the C–Cl bond can lead to chlorination of the substrate. Strongly absorbing solvents prevent photons from reaching the substrate. Hydrocarbon solvents absorb only at short wavelengths and are thus preferred for photochemical experiments requiring high energy photons. Solvents containing unsaturation absorb at longer wavelengths and can usefully filter out short wavelengths. For example, cyclohexane and acetone "cut off" (absorb strongly) at wavelengths shorter than 215 and 330 nm, respectively.
Photochemistry in combination with flow chemistry
Continuous flow photochemistry offers multiple advantages over batch photochemistry. Photochemical reactions are driven by the number of photons that are able to activate molecules causing the desired reaction. The large surface area to volume ratio of a microreactor maximizes the illumination, and at the same time allows for efficient cooling, which decreases the thermal side products.[7]
Principles
In the case of photochemical reactions, light provides the activation energy. Simplistically, light is one mechanism for providing the activation energy required for many reactions. If laser light is employed, it is possible to selectively excite a molecule so as to produce a desired electronic and vibrational state.[8] Equally, the emission from a particular state may be selectively monitored, providing a measure of the population of that state. If the chemical system is at low pressure, this enables scientists to observe the energy distribution of the products of a chemical reaction before the differences in energy have been smeared out and averaged by repeated collisions.
The absorption of a photon of light by a reactant molecule may also permit a reaction to occur not just by bringing the molecule to the necessary activation energy, but also by changing the symmetry of the molecule's electronic configuration, enabling an otherwise inaccessible reaction path, as described by the Woodward–Hoffmann selection rules. A 2+2 cycloaddition reaction is one example of a pericyclic reaction that can be analyzed using these rules or by the related frontier molecular orbital theory.
Some photochemical reactions are several orders of magnitude faster than thermal reactions; reactions as fast as 10−9 seconds and associated processes as fast as 10−15 seconds are often observed.
The photon can be absorbed directly by the reactant or by a photosensitizer, which absorbs the photon and transfers the energy to the reactant. The opposite process is called quenching when a photoexcited state is deactivated by a chemical reagent.
Most photochemical transformations occur through a series of simple steps known as primary photochemical processes. One common example of these processes is the excited state proton transfer.
Photochemical reactions
Examples of photochemical reactions
- Photosynthesis: plants use solar energy to convert carbon dioxide and water into glucose and oxygen.
- Human formation of vitamin D by exposure to sunlight.
- Bioluminescence: e.g. In fireflies, an enzyme in the abdomen catalyzes a reaction that produced light.[9]
- Polymerizations started by photoinitiators, which decompose upon absorbing light to produce the free radicals for radical polymerization.
- Photodegradation of many substances, e.g. polyvinyl chloride and Fp. Medicine bottles are often made with darkened glass to prevent the drugs from photodegradation.
- Photochemical rearrangements, e.g. photoisomerisation, hydrogen atom transfer and photochemical electrocyclic reactions.[10][11]
- Photodynamic therapy: light is used to destroy tumors by the action of singlet oxygen generated by photosensitized reactions of triplet oxygen. Typical photosensitizers include tetraphenylporphyrin and methylene blue. The resulting singlet oxygen is an aggressive oxidant, capable of converting C–H bonds into C–OH groups.
- Diazo printing process
- Photoresist technology, used in the production of microelectronic components.
- Vision is initiated by a photochemical reaction of rhodopsin.[12]
- Toray photochemical production of ε-caprolactame.[13]
- Photochemical production of artemisinin, anti-malaria drug.[14][15]
- Photoalkylation, used for the light-induced addition of alkyl groups to molecules.
- DNA: photodimerization leading to cyclobutane pyrimidine dimers.[16]
Organic photochemistry
Examples of photochemical organic reactions are electrocyclic reactions, radical reactions, photoisomerization and Norrish reactions.[17][18]
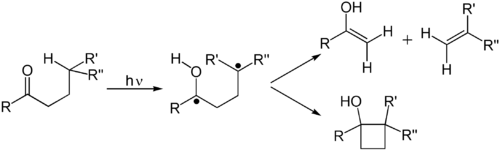
Alkenes undergo many important reactions that proceed via a photon-induced π to π* transition. The first electronic excited state of an alkene lack the π-bond, so that rotation about the C–C bond is rapid and the molecule engages in reactions not observed thermally. These reactions include cis-trans isomerization, cycloaddition to other (ground state) alkene to give cyclobutane derivatives. The cis-trans isomerization of a (poly)alkene is involved in retinal, a component of the machinery of vision. The dimerization of alkenes is relevant to the photodamage of DNA, where thymine dimers are observed upon illuminating DNA to UV radiation. Such dimers interfere with transcription. The beneficial effects of sunlight are associated with the photochemically induced retro-cyclization (decyclization) reaction of ergosterol to give vitamin D. In the DeMayo reaction, an alkene reacts with a 1,3-diketone reacts via its enol to yield a 1,5-diketone. Still another common photochemical reaction is Howard Zimmerman's di-π-methane rearrangement.
In an industrial application, about 100,000 tonnes of benzyl chloride are prepared annually by the gas-phase photochemical reaction of toluene with chlorine.[19] The light is absorbed by chlorine molecule, the low energy of this transition being indicated by the yellowish color of the gas. The photon induces homolysis of the Cl-Cl bond, and the resulting chlorine radical converts toluene to the benzyl radical:
- Cl2 + hν → 2 Cl·
- C6H5CH3 + Cl· → C6H5CH2· + HCl
- C6H5CH2· + Cl· → C6H5CH2Cl
Mercaptans can be produced by photochemical addition of hydrogen sulfide (H2S) to alpha olefins.
Inorganic and organometallic photochemistry
Coordination complexes and organometallic compounds are also photoreactive. These reactions can entail cis-trans isomerization. More commonly photoreactions result in dissociation of ligands, since the photon excites an electron on the metal to an orbital that is antibonding with respect to the ligands. Thus, metal carbonyls that resist thermal substitution undergo decarbonylation upon irradiation with UV light. UV-irradiation of a THF solution of molybdenum hexacarbonyl gives the THF complex, which is synthetically useful:
- Mo(CO)6 + THF → Mo(CO)5(THF) + CO
In a related reaction, photolysis of iron pentacarbonyl affords diiron nonacarbonyl (see figure):
- 2 Fe(CO)5 → Fe2(CO)9 + CO
Select photoreactive coordination complexes can undergo oxidation-reduction processes via single electron transfer. This electron transfer can occur within the inner or outer coordination sphere of the metal.[20]
Types of photochemical reactions
Here are some different types of photochemical reactions-
- Photo-dissociation: AB + hν → A* + B*
- Photo induced rearrangements, isomerization: A + hν → B
- Photo-Addition: A + B + hν → AB + C
- Photo-substitution: A + BC + hν → AB + C
- Photo-redox reaction: A + B + hν → A− + B+
Historical
Although bleaching has long been practiced, the first photochemical reaction was described by Trommsdorff in 1834.[21] He observed that crystals of the compound α-santonin when exposed to sunlight turned yellow and burst. In a 2007 study the reaction was described as a succession of three steps taking place within a single crystal.[22]
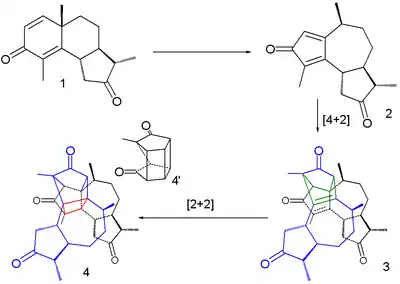
The first step is a rearrangement reaction to a cyclopentadienone intermediate 2, the second one a dimerization in a Diels–Alder reaction (3) and the third one an intramolecular [2+2]cycloaddition (4). The bursting effect is attributed to a large change in crystal volume on dimerization.
Specialized journals
Learned Societies
- Inter-American Photochemical Society
- European Photochemistry Association
- Asian and Oceanian Photochemistry Association
International conferences
- IUPAC SYmposium on Photochemistry (biennial)
- International Conference on Photochemitry (biennial)
The organization of these conferences is facilitated by the International Foundation for Photochemistry.[23]
See also
References
- IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "photochemistry". doi:10.1351/goldbook.P04588
- Glusac, Ksenija (2016). "What has light ever done for chemistry?". Nature Chemistry. 8 (8): 734–735. Bibcode:2016NatCh...8..734G. doi:10.1038/nchem.2582. PMID 27442273.
- J. Cadet and T. Douki Photochem. & Photobiol. Sci. 2018 (17) pp 1816-1841 DOI: 10.1039/c7pp00395a
- Calvert, J. G.; Pitts, J. N. Photochemistry. Wiley & Sons: New York, US, 1966. Congress Catalog number: 65-24288
- Photochemistry, website of William Reusch (Michigan State University), accessed 26 June 2016
- Wayne, C. E.; Wayne, R. P. Photochemistry, 1st ed.; Oxford University Press: Oxford, United Kingdom, reprinted 2005. ISBN 0-19-855886-4.
- Oelgemöller, Michael; Shvydkiv, Oksana (2011). "Recent Advances in Microflow Photochemistry". Molecules. 16 (9): 7522–7550. doi:10.3390/molecules16097522. PMC 6264405. PMID 21894087.
- Menzel, Jan P.; Noble, Benjamin B.; Lauer, Andrea; Coote, Michelle L.; Blinco, James P.; Barner-Kowollik, Christopher (2017). "Wavelength Dependence of Light-Induced Cycloadditions". Journal of the American Chemical Society. 139 (44): 15812–15820. doi:10.1021/jacs.7b08047. hdl:1885/209117. ISSN 0002-7863. PMID 29024596.
- Saunders, D. S. (2002-11-11). Insect Clocks, Third Edition. Elsevier Science. p. 179. ISBN 0444504079.
- Lefebvre, Corentin; Hoffmann, Norbert (2021-01-01), Török, Béla; Schäfer, Christian (eds.), "Chapter Eight – Photochemical rearrangements in organic synthesis and the concept of the photon as a traceless reagent", Nontraditional Activation Methods in Green and Sustainable Applications, Advances in Green and Sustainable Chemistry, Elsevier, pp. 283–328, doi:10.1016/b978-0-12-819009-8.00008-6, ISBN 978-0-12-819009-8, S2CID 234209169, retrieved 2022-01-24
- Lefebvre, Corentin; Fortier, Lucas; Hoffmann, Norbert (2020). "Photochemical Rearrangements in Heterocyclic Chemistry". European Journal of Organic Chemistry. 2020 (10): 1393–1404. doi:10.1002/ejoc.201901190. ISSN 1099-0690. S2CID 204117942.
- Dugave, Christophe (2006-10-06). Cis-trans Isomerization in Biochemistry. John Wiley & Sons. pp. 56. ISBN 9783527313044.
- Protti, Stefano; Fagnoni, Maurizio (2009). "The sunny side of chemistry: Green synthesis by solar light". Photochemical & Photobiological Sciences. 8 (11): 1499–516. doi:10.1039/B909128A. PMID 19862408. S2CID 9323784.
- Peplow, Mark (17 April 2013). "Sanofi launches malaria drug production". Chemistry World.
- Paddon, C. J.; Westfall, P. J.; Pitera, D. J.; Benjamin, K.; Fisher, K.; McPhee, D.; Leavell, M. D.; Tai, A.; Main, A.; Eng, D.; Polichuk, D. R. (2013). "High-level semi-synthetic production of the potent antimalarial artemisinin". Nature. 496 (7446): 528–532. Bibcode:2013Natur.496..528P. doi:10.1038/nature12051. ISSN 0028-0836. PMID 23575629.
- CYCLOBUTANE- TYPE PYRIMIDINE DIMERS IN POLYNUCLEOTIDES, R. B. Setlow, Science 1966 Vol. 153, p. 379, DOI: 10.1126/science.153.3734.379
- Klán, Petr; Wirz, Jakob (2009-03-23). Photochemistry of Organic Compounds: From Concepts to Practice. John Wiley & Sons. ISBN 978-1405190886.
- Turro, Nicholas J.; Ramamurthy, V.; Scaiano, Juan C. (2010). Modern Molecular Photochemistry of Organic Molecules. University Science Books. ISBN 978-1891389252.
- Rossberg, Manfred; Lendle, Wilhelm; Pfleiderer, Gerhard; Tögel, Adolf; Dreher, Eberhard-Ludwig; Langer, Ernst; Rassaerts, Heinz; Kleinschmidt, Peter; Strack, Heinz; Cook, Richard; Beck, Uwe; Lipper, Karl-August; Torkelson, Theodore R.; Löser, Eckhard; Beutel, Klaus K.; Mann, Trevor (2006). "Chlorinated Hydrocarbons". Ullmann's Encyclopedia of Industrial Chemistry. doi:10.1002/14356007.a06_233.pub2. ISBN 3527306730.
- Balzani, Vincenzo; Carassiti, Vittorio (1970). Photochemistry of Coordination Compounds. New York, New York: Academic Press, Inc. pp. 37–39. ISBN 9780120772506.
- Trommsdorff, Hermann (1834). "Ueber Santonin". Annalen der Pharmacie. 11 (2): 190–207. doi:10.1002/jlac.18340110207.
- Natarajan, Arunkumar; Tsai, C. K.; Khan, Saeed I.; McCarren, Patrick; Houk, K. N.; Garcia-Garibay, Miguel A. (2007). "The Photoarrangement of α-Santonin is a Single-Crystal-to-Single-Crystal Reaction: A Long Kept Secret in Solid-State Organic Chemistry Revealed". Journal of the American Chemical Society. 129 (32): 9846–9847. doi:10.1021/ja073189o. PMID 17645337.
- IUPAC Symposia on Photochemistry. A Brief History, S. Braslavsky, Chemistry International, March–April 2014.
Further reading
- Bowen, E. J., Chemical Aspects of Light. Oxford: The Clarendon Press, 1942. 2nd edition, 1946.
- Photochemistry
Branches of chemistry | |
|---|---|
| Analytical | |
| Theoretical | |
| Physical | |
| Inorganic | |
| Organic | |
| Biological | |
| Interdisciplinarity | |
| See also | |