Population structure (genetics)
Population structure (also called genetic structure and population stratification) is the presence of a systematic difference in allele frequencies between subpopulations. In a randomly mating (or panmictic) population, allele frequencies are expected to be roughly similar between groups. However, mating tends to be non-random to some degree, causing structure to arise. For example, a barrier like a river can separate two groups of the same species and make it difficult for potential mates to cross; if a mutation occurs, over many generations it can spread and become common in one subpopulation while being completely absent in the other.
Genetic variants do not necessarily cause observable changes in organisms, but can be correlated by coincidence because of population structure—a variant that is common in a population that has a high rate of disease may erroneously be thought to cause the disease. For this reason, population structure is a common confounding variable in medical genetics studies, and accounting for and controlling its effect is important in genome wide association studies (GWAS). By tracing the origins of structure, it is also possible to study the genetic ancestry of groups and individuals.
Description
The basic cause of population structure in sexually reproducing species is non-random mating between groups: if all individuals within a population mate randomly, then the allele frequencies should be similar between groups. Population structure commonly arises from physical separation by distance or barriers, like mountains and rivers, followed by genetic drift. Other causes include gene flow from migrations, population bottlenecks and expansions, founder effects, evolutionary pressure, random chance, and (in humans) cultural factors. Even in lieu of these factors, individuals tend to stay close to where they were born, which means that alleles will not be distributed at random with respect to the full range of the species.[1][2]
Measures
Population structure is a complex phenomenon and no single measure captures it entirely. Understanding a population's structure requires a combination of methods and measures.[3][4] Many statistical methods rely on simple population models in order to infer historical demographic changes, such as the presence of population bottlenecks, admixture events or population divergence times. Often these methods rely on the assumption of panmictia, or homogeneity in an ancestral population. Misspecification of such models, for instance by not taking into account the existence of structure in an ancestral population, can give rise to heavily biased parameter estimates.[5] Simulation studies show that historical population structure can even have genetic effects that can easily be misinterpreted as historical changes in population size, or the existence of admixture events, even when no such events occurred.[6]
Heterozygosity
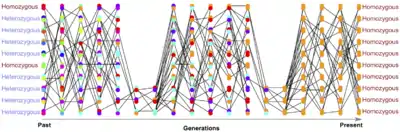
One of the results of population structure is a reduction in heterozygosity. When populations split, alleles have a higher chance of reaching fixation within subpopulations, especially if the subpopulations are small or have been isolated for long periods. This reduction in heterozygosity can be thought of as an extension of inbreeding, with individuals in subpopulations being more likely to share a recent common ancestor.[7] The scale is important — an individual with both parents born in the United Kingdom is not inbred relative to that country's population, but is more inbred than two humans selected from the entire world. This motivates the derivation of Wright's F-statistics (also called "fixation indices"), which measure inbreeding through observed versus expected heterozygosity.[8] For example, measures the inbreeding coefficient at a single locus for an individual relative to some subpopulation :[9]




Here, is the fraction of individuals in subpopulation that are heterozygous. Assuming there are two alleles, that occur at respective frequencies , it is expected that under random mating the subpopulation will have a heterozygosity rate of . Then:





Similarly, for the total population , we can define allowing us to compute the expected heterozygosity of subpopulation and the value as:[9]




If F is 0, then the allele frequencies between populations are identical, suggesting no structure. The theoretical maximum value of 1 is attained when an allele reaches total fixation, but most observed maximum values are far lower.[7] FST is one of the most common measures of population structure and there are several different formulations depending on the number of populations and the alleles of interest. Although it is sometimes used as a genetic distance between populations, it does not always satisfy the triangle inequality and thus is not a metric.[10] It also depends on within-population diversity, which makes interpretation and comparison difficult.[4]
Admixture inference
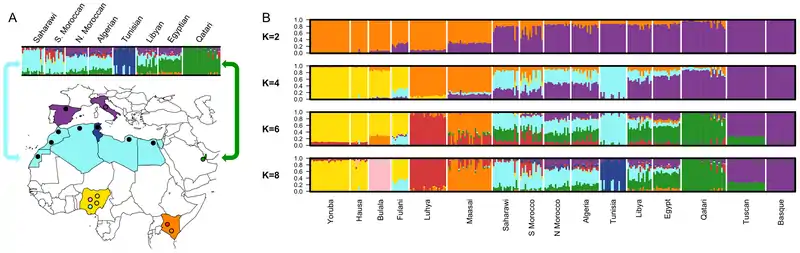
An individual's genotype can be modelled as an admixture between K discrete clusters of populations.[9] Each cluster is defined by the frequencies of its genotypes, and the contribution of a cluster to an individual's genotypes is measured via an estimator. In 2000, Jonathan K. Pritchard introduced the STRUCTURE algorithm to estimate these proportions via Markov chain Monte Carlo, modelling allele frequencies at each locus with a Dirichlet distribution.[11] Since then, algorithms (such as ADMIXTURE) have been developed using other estimation techniques.[12][13] Estimated proportions can be visualized using bar plots — each bar represents an individual, and is subdivided to represent the proportion of an individual's genetic ancestry from one of the K populations.[9]
Varying K can illustrate different scales of population structure; using a small K for the entire human population will subdivide people roughly by continent, while using large K will partition populations into finer subgroups.[9] Though clustering methods are popular, they are open to misinterpretation: for non-simulated data, there is never a "true" value of K, but rather an approximation considered useful for a given question.[3] They are sensitive to sampling strategies, sample size, and close relatives in data sets; there may be no discrete populations at all; and there may be hierarchical structure where subpopulations are nested.[3] Clusters may be admixed themselves,[9] and may not have a useful interpretation as source populations.[14]
Dimensionality reduction
Genetic data are high dimensional and dimensionality reduction techniques can capture population structure. Principal component analysis (PCA) was first applied in population genetics in 1978 by Cavalli-Sforza and colleagues and resurged with high-throughput sequencing.[9][17] Initially PCA was used on allele frequencies at known genetic markers for populations, though later it was found that by coding SNPs as integers (for example, as the number of non-reference alleles) and normalizing the values, PCA could be applied at the level of individuals.[13][18] One formulation considers individuals and bi-allelic SNPs. For each individual , the value at locus is is the number of non-reference alleles (one of ). If the allele frequency at is , then the resulting matrix of normalized genotypes has entries:[9]








PCA transforms data to maximize variance; given enough data, when each individual is visualized as point on a plot, discrete clusters can form.[13] Individuals with admixed ancestries will tend to fall between clusters, and when there is homogenous isolation by distance in the data, the top PC vectors will reflect geographic variation.[19][13] The eigenvectors generated by PCA can be explicitly written in terms of the mean coalescent times for pairs of individuals, making PCA useful for inference about the population histories of groups in a given sample. PCA cannot, however, distinguish between different processes that lead to the same mean coalescent times.[20]
Multidimensional scaling and discriminant analysis have been used to study differentiation, population assignment, and to analyze genetic distances.[21] Neighborhood graph approaches like t-distributed stochastic neighbor embedding (t-SNE) and uniform manifold approximation and projection (UMAP) can visualize continental and subcontinental structure in human data.[22][23] With larger datasets, UMAP better captures multiple scales of population structure; fine-scale patterns can be hidden or split with other methods, and these are of interest when the range of populations is diverse, when there are admixed populations, or when examining relationships between genotypes, phenotypes, and/or geography.[23][24] Variational autoencoders can generate artificial genotypes with structure representative of the input data, though they do not recreate linkage disequilibrium patterns.[25]
Demographic inference
Population structure is an important aspect of evolutionary and population genetics. Events like migrations and interactions between groups leave a genetic imprint on populations. Admixed populations will have haplotype chunks from their ancestral groups, which gradually shrink over time because of recombination. By exploiting this fact and matching shared haplotype chunks from individuals within a genetic dataset, researchers may trace and date the origins of population admixture and reconstruct historic events such as the rise and fall of empires, slave trades, colonialism, and population expansions.[26]
Role in genetic epidemiology
Population structure can be a problem for association studies, such as case-control studies, where the association between the trait of interest and locus could be incorrect. As an example, in a study population of Europeans and East Asians, an association study of chopstick usage may "discover" a gene in the Asian individuals that leads to chopstick use. However, this is a spurious relationship as the genetic variant is simply more common in Asians than in Europeans.[27] Also, actual genetic findings may be overlooked if the locus is less prevalent in the population where the case subjects are chosen. For this reason, it was common in the 1990s to use family-based data where the effect of population structure can easily be controlled for using methods such as the transmission disequilibrium test (TDT).[28]
Phenotypes (measurable traits), such as height or risk for heart disease, are the product of some combination of genes and environment. These traits can be predicted using polygenic scores, which seek to isolate and estimate the contribution of genetics to a trait by summing the effects of many individual genetic variants. To construct a score, researchers first enrol participants in an association study to estimate the contribution of each genetic variant. Then, they can use the estimated contributions of each genetic variant to calculate a score for the trait for an individual who was not in the original association study. If structure in the study population is correlated with environmental variation, then the polygenic score is no longer measuring the genetic component alone.[29]
Several methods can at least partially control for this confounding effect. The genomic control method was introduced in 1999 and is a relatively nonparametric method for controlling the inflation of test statistics.[30] It is also possible to use unlinked genetic markers to estimate each individual's ancestry proportions from some K subpopulations, which are assumed to be unstructured.[31] More recent approaches make use of principal component analysis (PCA), as demonstrated by Alkes Price and colleagues,[32] or by deriving a genetic relationship matrix (also called a kinship matrix) and including it in a linear mixed model (LMM).[33][34]
PCA and LMMs have become the most common methods to control for confounding from population structure. Though they are likely sufficient for avoiding false positives in association studies, they are still vulnerable to overestimating effect sizes of marginally associated variants and can substantially bias estimates of polygenic scores and trait heritability.[35][36] If environmental effects are related to a variant that exists in only one specific region (for example, a pollutant is found in only one city), it may not be possible to correct for this population structure effect at all.[29] For many traits, the role of structure is complex and not fully understood, and incorporating it into genetic studies remains a challenge and is an active area of research.[37]
References
- Cardon LR, Palmer LJ (February 2003). "Population stratification and spurious allelic association". Lancet. 361 (9357): 598–604. doi:10.1016/S0140-6736(03)12520-2. PMID 12598158. S2CID 14255234.
- McVean G (2001). "Population Structure" (PDF). Archived from the original (PDF) on 2018-11-23. Retrieved 2020-11-14.
- Lawson, Daniel J.; van Dorp, Lucy; Falush, Daniel (2018). "A tutorial on how not to over-interpret STRUCTURE and ADMIXTURE bar plots". Nature Communications. 9 (1): 3258. Bibcode:2018NatCo...9.3258L. doi:10.1038/s41467-018-05257-7. ISSN 2041-1723. PMC 6092366. PMID 30108219.
- Meirmans, Patrick G.; Hedrick, Philip W. (2010). "Assessing population structure:FST and related measures". Molecular Ecology Resources. 11 (1): 5–18. doi:10.1111/j.1755-0998.2010.02927.x. ISSN 1755-098X. PMID 21429096. S2CID 24403040.
- Scerri EM, Thomas MG, Manica A, Gunz P, Stock JT, Stringer C, et al. (August 2018). "Did Our Species Evolve in Subdivided Populations across Africa, and Why Does It Matter?". Trends in Ecology & Evolution. 33 (8): 582–594. doi:10.1016/j.tree.2018.05.005. PMC 6092560. PMID 30007846.
- Rodríguez W, Mazet O, Grusea S, Arredondo A, Corujo JM, Boitard S, Chikhi L (December 2018). "The IICR and the non-stationary structured coalescent: towards demographic inference with arbitrary changes in population structure". Heredity. 121 (6): 663–678. doi:10.1038/s41437-018-0148-0. PMC 6221895. PMID 30293985.
- Hartl, Daniel L.; Clark, Andrew G. (1997). Principles of population genetics (3rd ed.). Sunderland, MA: Sinauer Associates. pp. 111–163. ISBN 0-87893-306-9. OCLC 37481398.
- Wright, Sewall (1949). "The Genetical Structure of Populations". Annals of Eugenics. 15 (1): 323–354. doi:10.1111/j.1469-1809.1949.tb02451.x. ISSN 2050-1420. PMID 24540312.
- Coop, Graham (2019). Population and Quantitative Genetics. pp. 22–44.
- Arbisser, Ilana M.; Rosenberg, Noah A. (2020). "FST and the triangle inequality for biallelic markers". Theoretical Population Biology. 133: 117–129. doi:10.1016/j.tpb.2019.05.003. ISSN 0040-5809. PMC 8448291. PMID 31132375.
- Pritchard, Jonathan K; Stephens, Matthew; Donnelly, Peter (2000). "Inference of Population Structure Using Multilocus Genotype Data". Genetics. 155 (2): 945–959. doi:10.1093/genetics/155.2.945. ISSN 1943-2631. PMC 1461096. PMID 10835412.
- Alexander, D. H.; Novembre, J.; Lange, K. (2009). "Fast model-based estimation of ancestry in unrelated individuals". Genome Research. 19 (9): 1655–1664. doi:10.1101/gr.094052.109. ISSN 1088-9051. PMC 2752134. PMID 19648217.
- Novembre J, Ramachandran S (2011). "Perspectives on human population structure at the cusp of the sequencing era". Annu Rev Genomics Hum Genet. 12: 245–74. doi:10.1146/annurev-genom-090810-183123. PMID 21801023.
- Novembre, John (2016). "Pritchard, Stephens, and Donnelly on Population Structure". Genetics. 204 (2): 391–393. doi:10.1534/genetics.116.195164. ISSN 1943-2631. PMC 5068833. PMID 27729489.
- Henn BM, Botigué LR, Gravel S, Wang W, Brisbin A, Byrnes JK, Fadhlaoui-Zid K, Zalloua PA, Moreno-Estrada A, Bertranpetit J, Bustamante CD, Comas D (January 2012). "Genomic ancestry of North Africans supports back-to-Africa migrations". PLOS Genet. 8 (1): e1002397. doi:10.1371/journal.pgen.1002397. PMC 3257290. PMID 22253600.
- Wang C, Zöllner S, Rosenberg NA (August 2012). "A quantitative comparison of the similarity between genes and geography in worldwide human populations". PLOS Genet. 8 (8): e1002886. doi:10.1371/journal.pgen.1002886. PMC 3426559. PMID 22927824.
- Menozzi, P; Piazza, A; Cavalli-Sforza, L (1978). "Synthetic maps of human gene frequencies in Europeans". Science. 201 (4358): 786–792. Bibcode:1978Sci...201..786M. doi:10.1126/science.356262. ISSN 0036-8075. PMID 356262.
- Patterson N, Price AL, Reich D (December 2006). "Population structure and eigenanalysis". PLOS Genetics. 2 (12): e190. doi:10.1371/journal.pgen.0020190. PMC 1713260. PMID 17194218.
- Novembre, John; Johnson, Toby; Bryc, Katarzyna; Kutalik, Zoltán; Boyko, Adam R.; Auton, Adam; Indap, Amit; King, Karen S.; Bergmann, Sven; Nelson, Matthew R.; Stephens, Matthew; Bustamante, Carlos D. (2008). "Genes mirror geography within Europe". Nature. 456 (7218): 98–101. Bibcode:2008Natur.456...98N. doi:10.1038/nature07331. ISSN 0028-0836. PMC 2735096. PMID 18758442.
- McVean, Gil (2009). "A Genealogical Interpretation of Principal Components Analysis". PLOS Genetics. 5 (10): e1000686. doi:10.1371/journal.pgen.1000686. ISSN 1553-7404. PMC 2757795. PMID 19834557.
- Jombart T, Pontier D, Dufour AB (April 2009). "Genetic markers in the playground of multivariate analysis". Heredity (Edinb). 102 (4): 330–41. doi:10.1038/hdy.2008.130. PMID 19156164. S2CID 10739417.
- Li W, Cerise JE, Yang Y, Han H (August 2017). "Application of t-SNE to human genetic data". J Bioinform Comput Biol. 15 (4): 1750017. doi:10.1142/S0219720017500172. PMID 28718343.
- Diaz-Papkovich A, Anderson-Trocmé L, Ben-Eghan C, Gravel S (November 2019). "UMAP reveals cryptic population structure and phenotype heterogeneity in large genomic cohorts". PLOS Genet. 15 (11): e1008432. doi:10.1371/journal.pgen.1008432. PMC 6853336. PMID 31675358.
- Sakaue S, Hirata J, Kanai M, Suzuki K, Akiyama M, Lai Too C, Arayssi T, Hammoudeh M, Al Emadi S, Masri BK, Halabi H, Badsha H, Uthman IW, Saxena R, Padyukov L, Hirata M, Matsuda K, Murakami Y, Kamatani Y, Okada Y (March 2020). "Dimensionality reduction reveals fine-scale structure in the Japanese population with consequences for polygenic risk prediction". Nat Commun. 11 (1): 1569. Bibcode:2020NatCo..11.1569S. doi:10.1038/s41467-020-15194-z. PMC 7099015. PMID 32218440.
- Battey CJ, Coffing GC, Kern AD (January 2021). "Visualizing population structure with variational autoencoders". G3 (Bethesda). 11 (1). doi:10.1093/g3journal/jkaa036. PMC 8022710. PMID 33561250.
- Hellenthal G, Busby GB, Band G, Wilson JF, Capelli C, Falush D, Myers S (February 2014). "A genetic atlas of human admixture history". Science. 343 (6172): 747–751. Bibcode:2014Sci...343..747H. doi:10.1126/science.1243518. PMC 4209567. PMID 24531965.
- Hamer D, Sirota L (January 2000). "Beware the chopsticks gene". Molecular Psychiatry. 5 (1): 11–3. doi:10.1038/sj.mp.4000662. PMID 10673763. S2CID 9760182.
- Pritchard JK, Rosenberg NA (July 1999). "Use of unlinked genetic markers to detect population stratification in association studies". American Journal of Human Genetics. 65 (1): 220–8. doi:10.1086/302449. PMC 1378093. PMID 10364535.
- Blanc J, Berg JJ (December 2020). "How well can we separate genetics from the environment?". eLife. 9: e64948. doi:10.7554/eLife.64948. PMC 7758058. PMID 33355092.
- Devlin B, Roeder K (December 1999). "Genomic control for association studies". Biometrics. 55 (4): 997–1004. doi:10.1111/j.0006-341X.1999.00997.x. PMID 11315092. S2CID 6297807.
- Pritchard JK, Stephens M, Rosenberg NA, Donnelly P (July 2000). "Association mapping in structured populations". American Journal of Human Genetics. 67 (1): 170–81. doi:10.1086/302959. PMC 1287075. PMID 10827107.
- Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D (August 2006). "Principal components analysis corrects for stratification in genome-wide association studies". Nature Genetics. 38 (8): 904–9. doi:10.1038/ng1847. PMID 16862161. S2CID 8127858.
- Yu J, Pressoir G, Briggs WH, Vroh Bi I, Yamasaki M, Doebley JF, et al. (February 2006). "A unified mixed-model method for association mapping that accounts for multiple levels of relatedness". Nature Genetics. 38 (2): 203–8. doi:10.1038/ng1702. PMID 16380716. S2CID 8507433.
- Loh PR, Tucker G, Bulik-Sullivan BK, Vilhjálmsson BJ, Finucane HK, Salem RM, et al. (March 2015). "Efficient Bayesian mixed-model analysis increases association power in large cohorts". Nature Genetics. 47 (3): 284–90. doi:10.1038/ng.3190. PMC 4342297. PMID 25642633.
- Zaidi AA, Mathieson I (November 2020). Perry GH, Turchin MC, Martin P (eds.). "Demographic history mediates the effect of stratification on polygenic scores". eLife. 9: e61548. doi:10.7554/eLife.61548. PMC 7758063. PMID 33200985.
- Sohail M, Maier RM, Ganna A, Bloemendal A, Martin AR, Turchin MC, et al. (March 2019). Nordborg M, McCarthy MI, Barton NH, Hermisson J (eds.). "Polygenic adaptation on height is overestimated due to uncorrected stratification in genome-wide association studies". eLife. 8: e39702. doi:10.7554/eLife.39702. PMC 6428571. PMID 30895926.
- Lawson DJ, Davies NM, Haworth S, Ashraf B, Howe L, Crawford A, et al. (January 2020). "Is population structure in the genetic biobank era irrelevant, a challenge, or an opportunity?". Human Genetics. 139 (1): 23–41. doi:10.1007/s00439-019-02014-8. PMC 6942007. PMID 31030318.