Discovery and development of proton pump inhibitors
Proton pump inhibitors (PPIs) block the gastric hydrogen potassium ATPase (H+/K+ ATPase) and inhibit gastric acid secretion. These drugs have emerged as the treatment of choice for acid-related diseases, including gastroesophageal reflux disease (GERD) and peptic ulcer disease. PPIs also can bind to other types of proton pumps such as those that occur in cancer cells and are finding applications in the reduction of cancer cell acid efflux and reduction of chemotherapy drug resistance.
History
Evidence emerged by the end of the 1970s that the newly discovered proton pump (H+/K+ ATPase) in the secretory membrane of the parietal cell was the final step in acid secretion.[1] Literature from anaesthetic screenings led attention to the potential antiviral compound pyridylthioacetamide which after further examination pointed the focus on an anti-secretory compound with unknown mechanisms of action called timoprazole.[2][3][4] Timoprazole is a pyridylmethylsulfinyl benzimidazole and appealed due to its simple chemical structure and its surprisingly high level of anti-secretory activity.[5]
Optimization of substituted benzimidazoles and their antisecretory effects were studied on the newly discovered proton pump to obtain higher pKa values of the pyridine, thereby facilitating accumulation within the parietal cell and increasing the rate of acid-mediated conversion to the active mediate. As a result of such optimization the first proton pump inhibiting drug, omeprazole, was released on the market.[4][6] Other PPIs like lansoprazole and pantoprazole would follow in its footsteps, claiming their share of a flourishing market, after their own course of development.
Basic structure
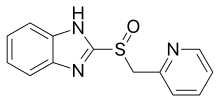
PPIs can be divided into two groups based on their basic structure. Although all members have a substituted pyridine part, one group has linked to various benzimidazoles, whereas the other has linked to a substituted imidazopyridine. All marketed PPIs (omeprazole, lansoprazole, pantoprazole) are in the benzimidazole group.
Proton pump inhibitors are prodrugs and their actual inhibitory form is somewhat controversial. In acidic solution, the sulfenic acid is isolated before reaction with one or more cysteines accessible from the luminar surface of the enzyme, a tetracyclic sulfenamide. This is a planar molecule thus any enantiomer of a PPI loses stereospecifity upon activation.[7]
The effectiveness of these drugs derives from two factors: their target, the H+/K+ ATPase which is responsible for the last step in acid secretion; therefore, their action on acid secretion is independent of the stimulus to acid secretion, of histamine, acetylcholine, or other yet to be discovered stimulants. In addition, their mechanism of action involves covalent binding of the activated drug to the enzyme, resulting in a duration of action that exceeds their plasma half-life.[7][8]
The gastric ATPase
Acid secretion by the human stomach results in a median diurnal pH of 1.4. This very large (>106-fold) H+ gradient is generated by the gastric H+/K+ ATPase which is an ATP-driven proton pump. Hydrolysis of one ATP molecule is used to catalyse the electroneutral exchange of two luminal potassium ions for two cytoplasmic protons through the gastric membrane.[9]
Structure
The proton pump, H+/K+ ATPase is a α,β-heterodimeric enzyme. The catalytic α subunit has ten transmembrane segments with a cluster of intramembranal carboxylic amino acids located in the middle of the transmembrane segments TM4, TM5, TM6 and TM8. The β subunit has one transmembrane segment with N terminus in cytoplasmic region. The extracellular domain of the β subunit contains six or seven N-linked glycosylation sites which is important for the enzyme assembly, maturation and sorting.[10]
Function
The ion transport is accomplished by cyclical conformational changes of the enzyme between its two main reaction states, E1 and E2. The cytoplasmic-open E1 and luminal-open E2 states have high affinity for H+ and K+.[9] The expulsion of the proton at 160 mM (pH 0.8) concentration results from movement of lysine 791 into the ion binding site in the E2P configuration.[10]
Discovery
In the year 1975, timoprazole was found to inhibit acid secretion irrespective of stimulus, extracellular or intracellular.[7] Studies on timoprazole revealed enlargement of the thyroid gland due to inhibition of iodine uptake as well as atrophy of the thymus gland. A literature search showed that some substituted mercapto-benzimidazoles had no effect on iodine uptake and introduction of such substituents into timoprazole resulted in an elimination of the toxic effects, without reducing the antisecretory effect.[6] A derivative of timoprazole, omeprazole, was discovered in 1979, and was the first of a new class of drug that control acid secretion in the stomach, a proton pump inhibitor (PPI).[11][12] Addition of 5-methoxy-substitution to the benzimidazole moiety of omeprazole was also made and gave the compound much more stability at neutral pH.[6] In 1980, an Investigational New Drug (IND) application was filed and omeprazole was taken into Phase III human trials in 1982.[6] A new approach for the treatment of acid-related diseases was introduced, and omeprazole was quickly shown to be clinically superior to the histamine H2 receptor antagonists, and was launched in 1988 as Losec in Europe, and in 1990 as Prilosec in the United States. In 1996, Losec became the world's biggest ever selling pharmaceutical, and by 2004 over 800 million patients had been treated with the drug worldwide. During the 1980s, about 40 other companies entered the PPIs area, but few achieved market success: Takeda with lansoprazole, Byk Gulden (now Nycomed) with pantoprazole, and Eisai with rabeprazole, all of which were analogues of omeprazole.[7][8]
Development
Pantoprazole
The story of pantoprazole's discovery is a good example of the stepwise development of PPIs. The main focus of modification of timoprazole was the benzimidazole part of its structure. Addition of a trifluoromethyl group to the benzimidazole moiety led to a series of very active compounds with varying solution-stability. In general fluoro substituents were found to block metabolism at the point where they were attached. Later the more balanced fluoroalkoxy substituent, instead of the highly lipophilic and strongly electron-withdrawing trifluoromethyl substituent, led to highly active compounds with supposed longer half-lives and higher solution stability.[5]
It was realized that activity was somehow linked to instability in solution and then came to the conclusion that the cyclic sulfenamides, formed in acidic conditions, were the active principle of the PPIs. Finally, it was understood that seemingly small alterations in the backbone of timoprazole led nowhere, and focus had to be centered on the substituents on the backbone. However, necessary intramolecular rearrangement of the benzimidazole into sulfenamide posed severe geometric constraints. Optimal compounds would be those that were stable at neutral pH but were quickly activated at low pH.[5]
A clear-cut design of active inhibitors was still not possible because in the complex multi-step chemistry the influence of a substituent on each step in the cascade could be different, and therefore not predictable for the overall rate of the prerequisite acid activation. Smith Kline and French, that entered into collaboration with Byk Gulden mid-1984, greatly assisted in determining criteria for further development. From 1985, the aim was to identify a compound with good stability at neutral pH, sustaining this higher level of stability down to pH 5 but being rapidly activateable at lower pHs, combined with a high level of H+/K+ ATPase inhibition.[13] From the numerous already synthesized and tested compounds that fulfilled these criteria the most promising candidates were pantoprazole and its salt, pantoprazole sodium.[5]
In 1986 pantoprazole sodium sesquihydrate was synthesized and from 1987 onwards the development of pantoprazole was switched to the sodium salt which is more stable and has better compatibility with other excipients used in the drug formulation.
Pantoprazole was identified after nearly seven years of research and registered for clinical use after a further seven years of development, and finally reached its first market in 1994 in Germany. During the course of the studies on pantoprazole, more than 650 PPIs had been synthesized and evaluated.[5] Pantoprazole obtained high selection criteria in its development process — especially concerning the favorable low potential for interaction with other drugs. Good solubility of pantoprazole and a very high solution stability allowed it to become the first marketed PPI for intravenous use in critical care patients.[5]
Esomeprazole
Omeprazole showed an inter-individual variability and therefore a significant number of patients with acid-related disorders required higher or multiple doses to achieve symptom relief and healing. Astra started a new research program in 1987 to identify a new analogue to omeprazole with less interpatient variability. Only one compound proved superior to omeprazole and that was the (S)-(−)-isomer, esomeprazole, which was developed as the magnesium salt. Esomeprazole magnesium (brand name Nexium) received its first approval in 2000 and provided more pronounced inhibition of acid secretion and less inter-patient variation compared to omeprazole. In 2004, Nexium had already been used to treat over 200 million patients.[7][8]
Benzimidazoles
- Omeprazole (brand names Losec, Prilosec, Zegerid, Ocid, Lomac, Omepral, Omez, Ultop, Ortanol, Gastrozol)
Omeprazole was the first PPI on the market, in 1988. It is a 1:1 racemate drug with a backbone structure of timoprazole, but substituted with two methoxy and two methyl groups. One of the methoxy group is at position 6 of the bensoimidazole and the other at position 4 of the pyridine and the methyl groups are at position 3 and 5 of the pyridine. Omeprazole is available as enteric-coated tablets, capsules, chewable tablets, powder for oral suspensions and powder for intravenous injection.
- Lansoprazole (brand names: Prevacid, Zoton, Inhibitol, Levant, Lupizole, Lancid, Lansoptol, Epicur)
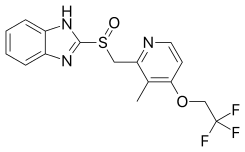
Lansoprazole was the second of the PPI drugs to reach the market, being launched in Europe in 1991 and the US in 1995. It has no substitutions at the benzimidazole but two substituents on the pyridine, methyl group at position 3 and a trifluoroethoxy group at position 4. The drug is a 1:1 racemate of the enantiomers dexlansoprazole and levolansoprazole. It is available in gastroresistant capsules and tablets as well as chewable tablets.
- Pantoprazole (brand names: Protonix, Somac, Pantoloc, Pantozol, Zurcal, Zentro, Pan, Nolpaza, Controloc, Sunpras)
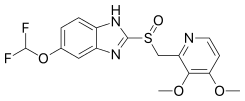
Pantoprazole was the third PPI and was introduced to the German market in 1994. It has a difluoroalkoxy sidegroup on the benzimidazole part and two methoxy groups in position 3 and 4 on the pyridine. Pantoprazole was first prepared in April 1985 by a small group of scale-up chemists. It is a dimethoxy-substituted pyridine bound to a fluoroalkoxy substituted benzimidazole.[5] Pantoprazole sodium is available as gastroresistant or delayed release tablets and as lyophilized powder for intravenous use.
- Rabeprazole (brand names: Zechin, Rabecid, Nzole-D, AcipHex, Pariet, Rabeloc, Zulbex, Ontime, Noflux)

Rabeprazole is a novel benzimidazole compound on market, since 1999 in USA. It is similar to lansoprazole in having no substituents on its benzimidazole part and a methyl group at site 3 on the pyridine, the only difference is the methoxypropoxy substitution at site 4 instead of the trifluoroethoxy group on lansoprazole. Rabeprazole is marketed as rabeprazole sodium salt. It is available as enteric-coated tablets.
- Esomeprazole (brand names: Nexium, Esotrex, Emanera, Neo-Zext)
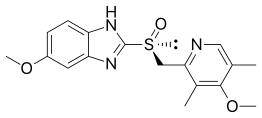
In 2001 esomeprazole was launched in USA, as a follow-up of omeprazoles patent. Esomeprazole is the (S)-(−)-enantiomer of omeprazole and provides higher bioavailability and improved efficacy, in terms of stomach acid control, over the (R)-(+)-enantiomer of omeprazole. In theory, by using pure esomeprazole the effects on the proton pump will be equal in all patients, eliminating the "poor metabolizer effect" of the racemate omeprazole. It is available as delayed-release capsules or tablets and as esomeprazole sodium for intravenous injection/infusion. Oral esomeprazole preparations are enteric-coated, due to the rapid degradation of the drug in the acidic condition of the stomach. This is achieved by formulating capsules using the multiple-unit pellet system. Although the (S)-(−)-isomer is more potent in humans, the (R)-(+)-isomer is more potent in testings of rats, while the enantiomers are equipotent in dogs.[14]
- Dexlansoprazole (brand names: Kapidex, Dexilant)

Dexlansoprazole was launched as a follow up of lansoprazole in 2009. Dexlansoprazole is an (R)-(+)-enantiomer of lansoprazole, marketed as Dexilant. After oral appliance of the racemic lansoprazole, the circulating drug is 80% dexlansoprazole. Moreover, both enantiomers have similar effects on the proton pump.[15] Consequently, the main advantage of Dexilant is not the fact that it is an enantiopure substance. The advantage is the pharmaceutical formulation of the drug, which is based on a dual release technology, with the first quick release producing a blood plasma peak concentration about one hour after application, and the second retarded release producing another peak about four hours later.[16]
Imidazopyridines
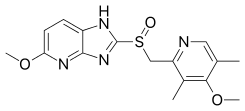
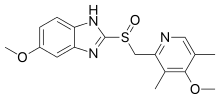
Tenatoprazole (TU-199), an imidazopyridine proton pump inhibitor, is a novel compound that has been designed as a new chemical entity with a substantially prolonged plasma half-life (7 hours), but otherwise has similar activity as other PPIs.[17]
The difference in the structural backbone of tenatoprazole compared to benzimidazole PPIs, is its imidazo[4,5-b]pyridine moiety, which reduces the rate of metabolism, allowing a longer plasma residence time but also decreases the pKa of the fused imidazole N as compared to the current PPIs.[18] Tenatoprazole has the same substituents as omeprazole, the methoxy groups at position 6 on the imidazopyridine and at position 4 on the pyridine part as well as two methyl groups at position 3 and 5 on the pyridine.
The bioavailability of tenatoprazole is double for the (S)-(−)-tenatoprazole sodium salt hydrate form when compared to the free form in dogs. This increased bioavailability is due to differences in the crystal structure and hydrophobic nature of the two forms, and therefore its more likely to be marketed as the pure (S)-(−)-enantiomer.
PPIs binding mode
The disulfide binding of the inhibitor takes place in the luminal sector of the H+/K+ ATPase were 2 mol of inhibitor is bound per 1 mol of active site H+/K+ ATPase.[19][20] All PPIs react with cysteine 813 in the loop between TM5 and TM6 on the H+/K+ ATPase, fixing the enzyme in the E2 configuration. Omeprazole reacts with cysteine 813 and 892. Rabeprazole binds to cysteine 813 and both 892 and 321. Lansoprazole reacts with cysteine 813 and cysteine 321, whereas pantoprazole and tenatoprazole react with cysteine 813 and 822.[18][21][22][23] Reaction with cysteine 822 confers a rather special property to the covalently inhibited enzyme, namely irreversibility to reducing agents. The likely first step is binding of the prodrug protonated on the pyridine of the compound with cysteine 813. Then the second proton is added with acid transport by the H+/K+ ATPase, and the compound is activated. Recent data suggest the hydrated sulfenic acid to be the reactive species forming directly from the mono-protonated benzimidazole bound on the surface of the pump.[7]
Saturation of the gastric ATPase
Even though consumption of food stimulates acid secretion and acid secretion activates PPIs, PPIs cannot inhibit all pumps. About 70% of pump enzyme is inhibited, as PPIs have a short half-life and not all pump enzymes are activated. It takes about 3 days to reach steady-state inhibition of acid secretion, as a balance is struck between covalent inhibition of active pumps, subsequent stimulation of inactive pumps after the drug has been eliminated from the blood, and de novo synthesis of new pumps.[8]
Clinical pharmacology
Although the drugs omeprazole, lansoprazole, pantoprazole, and rabeprazole share common structure and mode of action, each differs somewhat in its clinical pharmacology.[24] Differing pyridine and benzimidazole substituents result in small, but potentially significant different physical and chemical properties. Direct comparison of pantoprazole sodium with other anti-secretory drugs showed that it was significantly more effective than H2-receptor antagonists and either equivalent or better than other clinically used PPIs.[5] Another study states rabeprazole undergoes activation over a greater pH range than omeprazole, lansoprazole, and pantoprazole, and converts to the sulphenamide form more rapidly than any of these three drugs.[23] Most oral PPI preparations are enteric-coated, due to the rapid degradation of the drugs in the acidic conditions of the stomach. For example omeprazole is unstable in acid with a half-life of 2 min at pH 1–3, but is significantly more stable at pH 7 (half-life ca. 20 h). The acid protective coating prevents conversion to the active principle in the lumen of the stomach, which then will react with any available sulfhydryl group in food and will not penetrate to the lumen of the secretory canaliculus[10]
The oral bioavailability of PPIs is high; 77% for pantoprazole, 80–90% for lansoprazole and 89% for esomeprazole. All the PPIs except tenatoprazole are rapidly metabolized in the liver by CYP enzymes, mostly by CYP2C19 and CYP3A4. PPIs are sensitive to CYP enzymes and have different pharmacokinetic profiles. Studies comparing the efficacy of PPIs indicate that esomeprazole and tenatoprazole have stronger acid suppression, with a longer period of intragastric pH (pH > 4).[25][26][27][28][29]
Studies of the effect of tenatoprazole on acid secretion in in vivo animal models, such as pylorus-ligated rats and acute gastric fistula rats, demonstrated a 2- to 4-fold more potent inhibitory activity compared with omeprazole. A more potent inhibitory activity was also shown in several models of induced gastric lesions.[30] In Asian as well as Caucasian healthy subjects, tenatoprazole exhibited a seven-fold longer half-life than the existing H+/K+ ATPase inhibitors.[31] It is thus hypothesized that a longer half-life results in a more prolonged inhibition of gastric acid secretion, especially during the night. A strong relationship has been stated between the degree and duration of gastric acid inhibition, as measured by monitoring of the 24-hour intragastric pH in pharmacodynamic studies, and the rate of healing and symptom relief reported. A clinical study showed that nocturnal acid breakthrough duration was significantly shorter for 40 mg of tenatoprazole than for 40 mg of esomeprazole, with the conclusion that tenatoprazole was significantly more potent than esomeprazole during the night. Although, the therapeutic relevance of this pharmacological advantage deserves further study.[17]
PPIs have been used successfully in triple-therapy regiments with clarithromycin and amoxicillin for the eradication of Helicobacter pylori with no significant difference between different PPI-based regimens.[10]
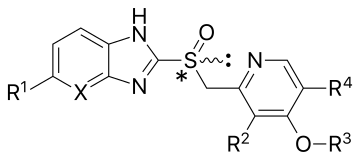
| Compound[18][21][22][23] | Substituents | Form | Cysteine binding | pKa | First approval | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| X | R1 | R2 | R3 | R4 | pKaa1 | pKaa1 | year | |||
| Omeprazole | CH | OCH3 | CH3 | CH3 | CH3 | Racemic mixture | 813 and 892 | 4.06 | 0.79 | 1989 in USA |
| Esomeprazole | CH | OCH3 | CH3 | CH3 | CH3 | (S)-(−)-enantiomer of omeprazole | 813 and 892 | 4.06 | 0.79 | 2001 in USA |
| Lansoprazole | CH | H | CH3 | CH2CF3 | H | Racemic mixture | 813 and 321 | 3.83 | 0.62 | 1991 in Europe |
| Dexlansoprazole | CH | H | CH3 | CH2CF3 | H | (R)-(+)-enantiomer of lansoprazole | 813 and 321 | 3.83 | 0.62 | 2009 in USA |
| Pantoprazole | CH | OCHF2 | OCH3 | CH3 | H | Racemic mixture | 813 and 822 | 3.83 | 0.11 | 1994 in Germany |
| Rabeprazole | CH | H | CH3 | (CH2)3OCH3 | H | Racemic mixture | 813, 892 and 321 | 4.53 | 0.62 | 1999 in USA |
| Tenatoprazole | N | OCH3 | CH3 | CH3 | CH3 | Racemic mixture | 813 and 822 | 4.04 | –0.12 | — |
Future research and new generations of PPIs
Potassium-competitive acid blockers or acid pump antagonists
Despite the fact that PPIs have revolutionized the treatment of GERD, there is still room for improvement in the speed of onset of acid suppression as well as mode of action that is independent of an acidic environment and also better inhibition of the proton pump.[8] Therefore, a new class of PPIs, potassium-competitive acid blockers (P-CABs) or acid pump antagonists (APAs), have been under development the past years and will most likely be the next generation of drugs that suppress gastric activity.[32] These new agents can in a reversible and competitive fashion inhibit the final step in the gastric acid secretion with respect to K+ binding to the parietal cell gastric H+/K+ ATPase. That is, they block the action of the H+/K+ ATPase by binding to or near the site of the K+ channel. Since the binding is competitive and reversible these agents have the potential to achieve faster inhibition of acid secretion and longer duration of action compared to PPIs, resulting in quicker symptom relief and healing.[33][34] The imidazopyridine-based compound SCH28080 was the prototype of this class, and turned out to be hepatotoxic.[35] Newer agents that are currently in development include CS-526, linaprazan, soraprazan and revaprazan in which the latter have reached clinical trials. Studies remain to determine whether these or other related compounds can become useful.[34][36] In June 2006, Yuhan obtained approval from the Korean FDA for the use of revaprazan (brand name Revanex) in the treatment of gastritis.[37] Vonoprazan is marketed in Japan.[38]
See also
References
- Forte, JG; Lee, HC (1977). "Gastric adenosine triphosphatases: A review of their possible role in HCl secretion". Gastroenterology. 73 (4 Pt 2): 921–6. doi:10.1016/S0016-5085(19)31737-8. PMID 20386.
- Snaeder, W. (1996). Drug prototypes and their exploitation. Wiley. pp. 414–5.
- Hemenway, Jeffrey N. (2007). "Case Study: Omeprazole (Prilosec)". Prodrugs. Biotechnology: Pharmaceutical Aspects. pp. 1313–21. doi:10.1007/978-0-387-49785-3_49. ISBN 978-0-387-49782-2.
- Olbe, L; Carlsson, E; Lindberg, P (February 2003). "A proton-pump inhibitor expedition: the case histories of omeprazole and esomeprazole". Nature Reviews. Drug Discovery. 2 (2): 132–9. doi:10.1038/nrd1010. PMID 12563304. S2CID 205474918.
- Senn-Bilfinger, Jörg; Sturm, Ernst (2006). "The Development of a New Proton-Pump Inhibitor: The Case History of Pantoprazole". Analogue-based Drug Discovery. pp. 115–36. doi:10.1002/3527608001.ch6. ISBN 978-3-527-60800-3.
- Lindberg, Per; Carlsson, Enar (2006). "Esomeprazole in the Framework of Proton-Pump Inhibitor Development". Analogue-based Drug Discovery. pp. 81–113. doi:10.1002/3527608001.ch5. ISBN 978-3-527-60800-3.
- Shin, Jai Moo; Munson, Keith; Vagin, Olga; Sachs, George (2008). "The gastric HK-ATPase: Structure, function, and inhibition". Pflügers Archiv: European Journal of Physiology. 457 (3): 609–22. doi:10.1007/s00424-008-0495-4. PMC 3079481. PMID 18536934.
- Sachs, George; Shin, Jai Moo; Vagin, Olga; Lambrecht, Nils; Yakubov, Iskandar; Munson, Keith (2007). "The Gastric H,K ATPase as a Drug Target". Journal of Clinical Gastroenterology. 41 (Suppl 2): S226–42. doi:10.1097/MCG.0b013e31803233b7. PMC 2860960. PMID 17575528.
- Abe, Kazuhiro; Tani, Kazutoshi; Nishizawa, Tomohiro; Fujiyoshi, Yoshinori (2009). "Inter-subunit interaction of gastric H+/K+ ATPase prevents reverse reaction of the transport cycle". The EMBO Journal. 28 (11): 1637–43. doi:10.1038/emboj.2009.102. PMC 2693145. PMID 19387495.
- Shin, Jai Moo; Sachs, George (2008). "Pharmacology of proton pump inhibitors". Current Gastroenterology Reports. 10 (6): 528–34. doi:10.1007/s11894-008-0098-4. PMC 2855237. PMID 19006606.
- Fellenius, Erik; Berglindh, Thomas; Sachs, George; Olbe, Lars; Elander, Berit; Sjöstrand, Sven-Erik; Wallmark, Björn (1981). "Substituted benzimidazoles inhibit gastric acid secretion by blocking (H+ + K+) ATPase". Nature. 290 (5802): 159–61. Bibcode:1981Natur.290..159F. doi:10.1038/290159a0. PMID 6259537. S2CID 4368190.
- Munson, Keith; Garcia, Rachel; Sachs, George (2005). "Inhibitor and Ion Binding Sites on the Gastric H,K-ATPase†". Biochemistry. 44 (14): 5267–84. doi:10.1021/bi047761p. PMID 15807521.
- Kohl, Bernhard; Sturm, Ernst; Senn-Bilfinger, Joerg; Simon, W. Alexander; Krueger, Uwe; Schaefer, Hartmann; Rainer, Georg; Figala, Volker; Klemm, Kurt (1992). "(H+, K+)-ATPase inhibiting 2-[(2-pyridylmethyl)sulfinyl]benzimidazoles. 4. A novel series of dimethoxypyridyl-substituted inhibitors with enhanced selectivity. The selection of pantoprazole as a clinical candidate". Journal of Medicinal Chemistry. 35 (6): 1049–57. doi:10.1021/jm00084a010. PMID 1313110.
- Silverman, Richard B. (2004). "Receptors". The organic chemistry of drug design and drug action (2nd ed.). Academic Press. p. 148. ISBN 9780126437324.
- Schubert-Zsilavecz, M, Wurglics, M, Neue Arzneimittel 2009
- Metz, D. C.; Vakily, M.; Dixit, T.; Mulford, D. (2009). "Review article: Dual delayed release formulation of dexlansoprazole MR, a novel approach to overcome the limitations of conventional single release proton pump inhibitor therapy". Alimentary Pharmacology & Therapeutics. 29 (9): 928–37. doi:10.1111/j.1365-2036.2009.03984.x. PMID 19298580. S2CID 29286087.
- Galmiche, J. P.; Bruley Des Varannes, S.; Ducrotte, P.; Sacher-Huvelin, S.; Vavasseur, F.; Taccoen, A.; Fiorentini, P.; Homerin, M. (2004). "Tenatoprazole, a novel proton pump inhibitor with a prolonged plasma half-life: Effects on intragastric pH and comparison with esomeprazole in healthy volunteers". Alimentary Pharmacology and Therapeutics. 19 (6): 655–62. doi:10.1111/j.1365-2036.2004.01893.x. PMID 15023167.
- Shin, Jai Moo; Homerin, Michel; Domagala, Florence; Ficheux, Hervé; Sachs, George (2006). "Characterization of the inhibitory activity of tenatoprazole on the gastric H+,K+-ATPase in vitro and in vivo". Biochemical Pharmacology. 71 (6): 837–49. doi:10.1016/j.bcp.2005.11.030. PMID 16405921.
- Kaminski, James J.; Doweyko, Arthur M. (1997). "Antiulcer Agents. 6. Analysis of the in Vitro Biochemical and in Vivo Gastric Antisecretory Activity of Substituted Imidazo[1,2-a]pyridines and Related Analogues Using Comparative Molecular Field Analysis and Hypothetical Active Site Lattice Methodologies". Journal of Medicinal Chemistry. 40 (4): 427–36. doi:10.1021/jm950700s. PMID 9046332.
- Lindberg, Per; Brändström, Arne; Wallmark, Björn; Mattsson, Hillevi; Rikner, Leif; Hoffmann, Kurt-Jürgen (1990). "Omeprazole: The first proton pump inhibitor". Medicinal Research Reviews. 10 (1): 1–54. doi:10.1002/med.2610100102. PMID 2404184. S2CID 8637440.
- J M Shin; Sachs, G (1994-03-25). "Identification of a region of the H,K-ATPase alpha subunit associated with the beta subunit". Journal of Biological Chemistry. 269 (12): 8642–6. doi:10.1016/S0021-9258(17)37015-1. PMID 8132592.
- Shin, Jai Moo; Besancon, Marie; Simon, Alexander; Sachs, George (1993). "The site of action of pantoprazole in the gastric H+/K+-ATPase". Biochimica et Biophysica Acta (BBA) - Biomembranes. 1148 (2): 223–33. doi:10.1016/0005-2736(93)90133-K. PMID 8389196.
- Primi, M.P.; Bueno, L.; Baumer, P.; Berard, H.; Lecomte, J.M. (1999). "Racecadotril demonstrates intestinal antisecretory activity in vivo". Alimentary Pharmacology and Therapeutics. 13 (Suppl 6): 3–7. doi:10.1046/j.1365-2036.13.s6.3.x. PMID 10646045. S2CID 25832405.
- Horn, John (2000). "The proton-pump inhibitors: Similarities and differences". Clinical Therapeutics. 22 (3): 266–80, discussion 265. doi:10.1016/S0149-2918(00)80032-6. PMID 10963283.
- Jorgensen, Peter L.; Håkansson, Kjell O.; Karlish, Steven J. D. (2003). "Structure and mechanism of Na, K-ATPase: functional sites and their interactions". Annual Review of Physiology. 65: 817–49. doi:10.1146/annurev.physiol.65.092101.142558. PMID 12524462. S2CID 334802.
- Blostein, R.; Dunbar, L; Mense, M; Scanzano, R; Wilczynska, A; Caplan, MJ (1999). "Cation Selectivity of Gastric H,K-ATPase and Na,K-ATPase Chimeras". Journal of Biological Chemistry. 274 (26): 18374–81. doi:10.1074/jbc.274.26.18374. PMID 10373442.
- McIntosh, D. B.; Clausen, JD; Woolley, DG; MacLennan, DH; Vilsen, B; Andersen, JP (2004). "Roles of Conserved P Domain Residues and Mg2+ in ATP Binding in the Ground and Ca2+-activated States of Sarcoplasmic Reticulum Ca2+-ATPase". Journal of Biological Chemistry. 279 (31): 32515–23. doi:10.1074/jbc.M403242200. PMID 15133025.
- Pont, JAN Joep H. H. M.; Swarts, Herman G. P.; Willems, Peter H. G. M.; Koenderink, JAN B. (2003). "The E1/E2-Preference of Gastric H,K-ATPase Mutants". Annals of the New York Academy of Sciences. 986 (1): 175–82. Bibcode:2003NYASA.986..175P. doi:10.1111/j.1749-6632.2003.tb07157.x. PMID 12763793. S2CID 35413255.
- Reuben, Michael A.; Lasater, Linda S.; Sachs, George (1990). "Characterization of a β Subunit of the Gastric H+/K+ Transporting ATPase". Proceedings of the National Academy of Sciences. 87 (17): 6767–71. Bibcode:1990PNAS...87.6767R. doi:10.1073/pnas.87.17.6767. JSTOR 2355381. PMC 54618. PMID 2168558.
- Uchiyama, K.; Wakatsuki, D.; Kakinoki, B.; Takeuchi, Y.; Araki, T.; Morinaka, Y. (1999). "The Long-lasting Effect of TU-199, a Novel H+,K+-ATPase Inhibitor, on Gastric Acid Secretion in Dogs". Journal of Pharmacy and Pharmacology. 51 (4): 457–64. doi:10.1211/0022357991772510. PMID 10385219. S2CID 41561851.
- Domagala, Florence; Ficheux, Herve (2003). "Pharmacokinetics of tenatoprazole, a novel proton pump inhibitor, in healthy male Caucasian volunteers". Gastroenterology. 124 (4, Supplement 1): A231. doi:10.1016/S0016-5085(03)81159-9.
- Scarpignato, C; Hunt, R (2008). "Proton pump inhibitors: The beginning of the end or the end of the beginning?". Current Opinion in Pharmacology. 8 (6): 677–84. doi:10.1016/j.coph.2008.09.004. PMID 18840545.
- Shin, J. M. & Sachs, G. (2009). "Long lasting inhibitors of the gastric H,K-ATPase". Expert Review of Clinical Pharmacology. 2 (5): 461–468. doi:10.1586/ecp.09.33. PMC 2995460. PMID 21132072.
- Nayana, M. Ravi Shashi; Sekhar, Y. Nataraja; Nandyala, Haritha; Muttineni, Ravikumar; Bairy, Santosh Kumar; Singh, Kriti; Mahmood, S.K. (2008). "Insight into the structural requirements of proton pump inhibitors based on CoMFA and CoMSIA studies". Journal of Molecular Graphics and Modelling. 27 (3): 233–43. doi:10.1016/j.jmgm.2008.04.012. PMID 18676164.
- Ravinder Reddy, B; Basavaraja, H S; Shivaprasad LV J, S. "Reversible Proton Pump Inhibitors: A Superior Edge — Features". Pharmabiz.com. Saffron Media Pvt. Ltd I. Retrieved 7 December 2015.
- Nelson, Wendel L (2008). "Antihistamines and Related Antiallergic and Antiulcer Agents". In Lemke, Thomas L.; Williams, David A. (eds.). Foye's Principles of Medicinal Chemistry (6th ed.). pp. 1004–27. ISBN 978-0-7817-6879-5.
- "Revaprazan Yuhan registered, South Korea (gastritis)". R & D Focus Drug News. September 25, 2006. Archived from the original on April 29, 2014.
- Garnock-Jones KP (2015). "Vonoprazan: first global approval". Drugs. 75 (4): 439–43. doi:10.1007/s40265-015-0368-z. PMID 25744862. S2CID 43293048.
Major chemical drug groups – based upon the Anatomical Therapeutic Chemical Classification System | |
|---|---|
| gastrointestinal tract / metabolism (A) | |
| blood and blood forming organs (B) | |
| cardiovascular system (C) | |
| skin (D) | |
| genitourinary system (G) | |
| endocrine system (H) | |
| infections and infestations (J, P, QI) | |
| malignant disease (L01–L02) | |
| immune disease (L03–L04) | |
| muscles, bones, and joints (M) | |
| brain and nervous system (N) |
|
| respiratory system (R) | |
| sensory organs (S) | |
| other ATC (V) | |
| |