Progeria
Progeria is a specific type of progeroid syndrome, also known as Hutchinson–Gilford syndrome. A single gene mutation is responsible for progeria. The gene, known as lamin A (LMNA), makes a protein necessary for holding the nucleus of the cell together. When this gene gets mutated, an abnormal form of lamin A protein called progerin is produced. Progeroid syndromes are a group of diseases that causes individuals to age faster than usual, leading to them appearing older than they actually are. Patients born with progeria typically live to an age of mid-teens to early twenties.[8][9]
| Progeria | |
|---|---|
| Other names | Hutchinson–Gilford progeria syndrome (HGPS),[1][2] progeria syndrome,[2] Joseph Syndrome |
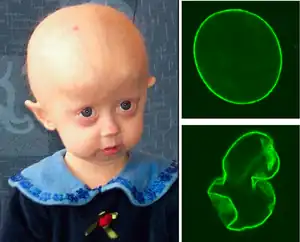 | |
| A young girl with progeria (left). A healthy cell nucleus (right, top) and a progeric cell nucleus (right, bottom). | |
| Pronunciation | |
| Specialty | Medical genetics |
| Symptoms | Growth delay, short height, small face, hair loss |
| Complications | Heart disease, stroke, hip dislocations[5] |
| Usual onset | 9–24 months[5] |
| Causes | Genetic[5] |
| Diagnostic method | Based on symptoms, genetic tests[5] |
| Differential diagnosis | Hallermann–Streiff syndrome, Gottron's syndrome, Wiedemann–Rautenstrauch syndrome[5] |
| Treatment | Mostly symptomatic[5] |
| Medication | Lonafarnib[6][7] |
| Prognosis | Average age of death is 15 years |
| Frequency | Rare: 1 in 18 million[5] |
Severe cardiovascular complications usually develop by puberty, resulting in death.
Signs and symptoms
Children with progeria usually develop the first symptoms during their first few months of life. The earliest symptoms may include a failure to thrive and a localized scleroderma-like skin condition. As a child ages past infancy, additional conditions become apparent, usually around 18–24 months. Limited growth, full-body alopecia (hair loss), and a distinctive appearance (a small face with a shallow, recessed jaw and a pinched nose) are all characteristics of progeria.[5] Signs and symptoms of this progressive disease tend to become more marked as the child ages. Later, the condition causes wrinkled skin, kidney failure, loss of eyesight, and atherosclerosis and other cardiovascular problems.[10] Scleroderma, a hardening and tightening of the skin on trunk and extremities of the body, is prevalent. People diagnosed with this disorder usually have small, fragile bodies, like those of older adults. The head is usually large relative to the body, with a narrow, wrinkled face and a beak nose. Prominent scalp veins are noticeable (made more obvious by alopecia), as well as prominent eyes. Musculoskeletal degeneration causes loss of body fat and muscle, stiff joints, hip dislocations, and other symptoms generally absent in the non-elderly population. Individuals usually retain typical mental and motor function.
Pathophysiology
Hutchinson-Gilford syndrome (HGPS) is an extremely rare autosomal dominant genetic disorder in which symptoms resembling aspects of aging are manifested at an early age.[11] Its occurrence is usually the result of a sporadic germline mutation; although HGPS is genetically dominant, people rarely live long enough to have children, preventing them from passing the disorder on in a hereditary manner.[12]
HPGS is caused by mutations that weaken the structure of the cell nucleus, making normal cell division difficult. The histone mark H4K20me3 is involved and caused by de novo mutations that occur in a gene that encodes lamin A. Lamin A is made but is not processed properly. This poor processing creates an abnormal nuclear morphology and disorganized heterochromatin. Patients also do not have appropriate DNA repair, and they also have increased genomic instability.[13]
In normal conditions, the LMNA gene codes for a structural protein called prelamin A, which undergoes a series of processing steps before attaining its final form, called lamin A.[14] Prelamin A contains a "CAAX" where C is a cysteine, A an aliphatic amino acid, and X any amino acid. This motif at the carboxyl-termini of proteins triggers three sequential enzymatic modifications. First, protein farnesyltransferase catalyzes the addition of a farnesyl moiety to the cysteine. Second, an endoprotease that recognizes the farnesylated protein catalyzes the peptide bond's cleavage between the cysteine and -aaX. In the third step, isoprenylcysteine carboxyl methyltransferase catalyzes methylation of the carboxyl-terminal farnesyl cysteine. The farnesylated and methylated protein is transported through a nuclear pore to the interior of the nucleus. Once in the nucleus, the protein is cleaved by a protease called zinc metallopeptidase STE24 (ZMPSTE24), which removes the last 15 amino acids, which includes the farnesylated cysteine. After cleavage by the protease, prelamin A is referred to as lamin A. In most mammalian cells, lamin A, along with lamin B1, lamin B2, and lamin C, makes up the nuclear lamina, which provides shape and stability to the inner nuclear envelope. Before the late 20th century, research on progeria yielded very little information about the syndrome. In 2003, the cause of progeria was discovered to be a point mutation in position 1824 of the LMNA gene, which replaces a cytosine with thymine.[15] This mutation creates a 5' cryptic splice site within exon 11, resulting in a shorter than normal mRNA transcript. When this shorter mRNA is translated into protein, it produces an abnormal variant of the prelamin A protein, referred to as progerin. Progerin's farnesyl group cannot be removed because the ZMPSTE24 cleavage site is lacking from progerin, so the abnormal protein is permanently attached to the nuclear rim. One result is that the nuclear lamina does not provide the nuclear envelope with enough structural support, causing it to take on an abnormal shape.[16] Since the support that the nuclear lamina normally provides is necessary for the organizing of chromatin during mitosis, weakening of the nuclear lamina limits the ability of the cell to divide.[17] However, defective cell division is unlikely to be the main defect leading to progeria, particularly because children develop normally without any signs of disease until about one year of age. Farnesylated prelamin A variants also lead to defective DNA repair, which may play a role in the development of progeria.[18] Progerin expression also leads to defects in the establishment of fibroblast cell polarity, which is also seen in physiological aging.[19]
To date over 1,400 SNPs in the LMNA gene are known.[20] They can manifest as changes in mRNA, splicing, or protein amino acid sequence (e.g. Arg471Cys,[21] Arg482Gln,[22] Arg527Leu,[23] Arg527Cys,[24] Ala529Val[25]).
Progerin may also play a role in normal human aging, since its production is activated in typical senescent cells.[17]
Unlike other "accelerated aging diseases" (such as Werner syndrome, Cockayne syndrome or xeroderma pigmentosum), progeria may not be directly caused by defective DNA repair. These diseases each cause changes in a few specific aspects of aging, but never in every aspect at once, so they are often called "segmental progerias".[26]
A 2003 report in Nature[27] said that progeria may be a de novo dominant trait. It develops during cell division in a newly conceived zygote or in the gametes of one of the parents. It is caused by mutations in the LMNA (lamin A protein) gene on chromosome 1; the mutated form of lamin A is commonly known as progerin. One of the authors, Leslie Gordon, was a physician who did not know anything about progeria until her own son, Sam, was diagnosed at 22 months. Gordon and her husband, pediatrician Scott Berns, founded the Progeria Research Foundation.[28]
Lamin A
Lamin A is a major component of a protein scaffold on the inner edge of the nucleus called the nuclear lamina that helps organize nuclear processes such as RNA and DNA synthesis.
Prelamin A contains a CAAX box at the C-terminus of the protein (where C is a cysteine and A is any aliphatic amino acids). This ensures that the cysteine is farnesylated and allows prelamin A to bind membranes, specifically the nuclear membrane. After prelamin A has been localized to the cell nuclear membrane, the C-terminal amino acids, including the farnesylated cysteine, are cleaved off by a specific protease. The resulting protein, now lamin A, is no longer membrane-bound and carries out functions inside the nucleus.
In HGPS, the recognition site that the enzyme requires for cleavage of prelamin A to lamin A is mutated. Lamin A cannot be produced, and prelamin A builds up on the nuclear membrane, causing a characteristic nuclear blebbing.[29] This results in the symptoms of progeria, although the relationship between the misshapen nucleus and the symptoms is not known.
A study that compared HGPS patient cells with the skin cells from young and elderly normal human subjects found similar defects in the HGPS and elderly cells, including down-regulation of certain nuclear proteins, increased DNA damage, and demethylation of histone, leading to reduced heterochromatin.[30] Nematodes over their lifespan show progressive lamin changes comparable to HGPS in all cells but neurons and gametes.[31] These studies suggest that lamin A defects are associated with normal aging.[30][32]
Mitochondria
The presence of progerin also leads to the accumulation of dysfunctional mitochondria within the cell. These mitochondria are characterized by a swollen morphology, caused by a condensation of mtDNA and TFAM into the mitochondria, which is driven by a severe mitochondrial dysfunction (low mitochondrial membrane potential, low ATP production, low respiration capacity and high ROS production).[33][34][35] Therefore, contributing substantially to the senescence phenotype. Although, the explanation for this defective-mitochondria accumulation in progeria is about to be elucidated, it has been proposed that low PGC1-α expression[33][34][36] (important for mitochondrial biogenesis, maintenance and function) along with low LAMP2 protein level and lysosome number (both important for mitophagy: the degradation of defective mitochondria pathway),[33] could be implicated.
Diagnosis
Skin changes, abnormal growth, and loss of hair occur. These symptoms normally start appearing by one year of age. A genetic test for LMNA mutations can confirm the diagnosis of progeria.[37][38] Prior to the advent of the genetic test, misdiagnosis was common.[38]
Treatment
In November 2020, the U.S. Food and Drug Administration approved lonafarnib, which helps prevent buildup of defective progerin and similar proteins.[39] A clinical trial in 2018 points to significantly lower mortality rates – treatment with lonafarnib alone compared with no treatment (3.7% vs. 33.3%) – at a median post-trial follow-up time span of 2.2 years.[40] The drug, given orphan drug status and Pediatric Disease Priority Review Voucher, is taken twice daily in the form of capsules and may cost US$650,000 per year, making it prohibitive for the vast majority of families. It is unclear how it will be covered by health insurance in the United States. Common side effects of the drug include "nausea, vomiting, diarrhea, infections, decreased appetite, and fatigue".[12]
Other treatment options have focused on reducing complications (such as cardiovascular disease) with coronary artery bypass surgery and low-dose acetylsalicylic acid.[41]
Growth hormone treatment has been attempted.[42] The use of Morpholinos has also been attempted in mice and cell cultures in order to reduce progerin production. Antisense Morpholino oligonucleotides specifically directed against the mutated exon 11–exon 12 junction in the mutated pre-mRNAs were used.[43]
A type of anticancer drug, the farnesyltransferase inhibitors (FTIs), has been proposed, but their use has been mostly limited to animal models.[44] A Phase II clinical trial using the FTI lonafarnib began in May 2007.[45] In studies on the cells another anti-cancer drug, rapamycin, caused removal of progerin from the nuclear membrane through autophagy.[16][46] It has been proved that pravastatin and zoledronate are effective drugs when it comes to the blocking of farnesyl group production.
Farnesyltransferase inhibitors (FTIs) are drugs that inhibit the activity of an enzyme needed to make a link between progerin proteins and farnesyl groups. This link generates the permanent attachment of the progerin to the nuclear rim. In progeria, cellular damage can occur because that attachment occurs, and the nucleus is not in a normal state. Lonafarnib is an FTI, which means it can avoid this link, so progerin can not remain attached to the nucleus rim, and it now has a more normal state.
Studies of sirolimus, an mTOR Inhibitor, demonstrate that it can minimize the phenotypic effects of progeria fibroblasts. Other observed consequences of its use are abolishing nuclear blebbing, degradation of progerin in affected cells, and reducing insoluble progerin aggregates formation. These results have been observed only in vitro and are not the results of any clinical trial, although it is believed that the treatment might benefit HGPS patients.[16]
Recently, it has been demonstrated that the CRM1 protein (a key component of the nuclear export machinery in mammalian) is upregulated in HGPS cells, which drives to the abnormal localization of NES containing proteins from the nucleus to the cytoplasm.[47] Moreover, the inhibition of CRM1 in HGPS alleviates the associated-senescence phenotype[47] as well as the mitochondrial function (an important determinant in senescence) and lysosome content.[33] These results are under in vivo validation with selinexor (a more suitable CRM1 inhibitor for human use[48]).
Prognosis
As there is no known cure, few people with progeria exceed 13 years of age.[49] At least 90 percent of patients die from complications of atherosclerosis, such as heart attack or stroke.[50]
Mental development is not adversely affected; in fact, intelligence tends to be average to above average.[51] With respect to the features of aging that progeria appears to manifest, the development of symptoms is comparable to aging at a rate eight to ten times faster than normal. With respect to those that progeria does not exhibit, patients show no neurodegeneration or cancer predisposition. They also do not develop conditions that are commonly associated with accumulation of damage, such as cataracts (caused by UV exposure) and osteoarthritis.[37]
Although there may not be any successful treatments for progeria itself, there are treatments for the problems it causes, such as arthritic, respiratory, and cardiovascular problems. People with progeria have normal reproductive development, and there are known cases of women with progeria who delivered healthy offspring.[52]
Epidemiology
A study from the Netherlands has shown an incidence of 1 in 20 million births.[53] According to the Progeria Research Foundation, as of September 2020, there are 179 known cases in the world, in 53 countries; 18 of the cases were identified in the United States.[54][12] Hundreds of cases have been reported in medical history since 1886.[55][56][57] However, the Progeria Research Foundation believes there may be as many as 150 undiagnosed cases worldwide.[58]
There have been only two cases in which a healthy person was known to carry the LMNA mutation that causes progeria.[59] One family from India had four of six children with progeria.[60]
Research
Mouse model
A mouse model of progeria exists, though in the mouse, the LMNA prelamin A is not mutated. Instead, ZMPSTE24, the specific protease that is required to remove the C-terminus of prelamin A, is missing. Both cases result in the buildup of farnesylated prelamin A on the nuclear membrane and in the characteristic nuclear LMNA blebbing.
In 2020 BASE editing was used in a mouse model to target the LMNA gene mutation that causes the progerin protein instead of the healthy Lamin A[61][62] while in 2023 a study designed a peptide that prevented progerin from binding to BubR1[63] which is known to regulate aging in mice.[64]
DNA repair
Repair of DNA double-strand breaks can occur by either of two processes, non-homologous end joining (NHEJ) or homologous recombination (HR). A-type lamins promote genetic stability by maintaining levels of proteins that have key roles in NHEJ and HR.[65] Mouse cells deficient for maturation of prelamin A show increased DNA damage and chromosome aberrations and have increased sensitivity to DNA damaging agents.[18] In progeria, the inability to adequately repair DNA damages due to defective A-type lamin may cause aspects of premature aging[66] (also see DNA damage theory of aging).
Epigenetic clock analysis of human HGPS
Fibroblast samples from children with progeria syndrome exhibit accelerated epigenetic aging effects according to the epigenetic clock for skin and blood samples.[67]
History
Progeria was first described in 1886 by Jonathan Hutchinson.[68] It was also described independently in 1897 by Hastings Gilford.[69] The condition was later named Hutchinson–Gilford progeria syndrome. Scientists are interested in progeria partly because it might reveal clues about the normal process of aging.[70][59][71]
Society and culture
Notable cases
Yan Hui, a student of Confucius, aged rapidly and died at a young age, appearing as an old man by his late 20s. He may be one of the earliest potential examples of progeria in history.[73]
In 1987, fifteen-year-old Mickey Hays, who had progeria, appeared along with Jack Elam in the documentary I Am Not a Freak.[74] Elam and Hays first met during the filming of the 1986 film The Aurora Encounter,[75] in which Hays was cast as an alien. The friendship that developed lasted until Hays died in 1992, on his 20th birthday. Elam said, "You know I've met a lot of people, but I've never met anybody that got next to me like Mickey."
Harold Kushner, who among other things wrote the book When Bad Things Happen to Good People, had a son, Aaron, who died at the age of 14 in 1977 of progeria.[76]
Margaret Casey, a 29-year-old woman with progeria who was then believed to be the oldest survivor of the premature aging disease, died on Sunday, May 26, 1985. Casey, a freelance artist, was admitted to Yale-New Haven Hospital on the night of May 25 with respiratory problems, which caused her death.[77]
Sam Berns was an American activist with the disease. He was the subject of the HBO documentary Life According to Sam. Berns also gave a TEDx talk titled My Philosophy for a Happy Life on December 13, 2013.[78]
Hayley Okines was an English progeria patient who spread awareness of the condition.[79]
Leon Botha, the South African painter and DJ who was known, among other things, for his work with the hip-hop duo Die Antwoord, lived with progeria.[80] He died in 2011, aged 26.
Tiffany Wedekind of Columbus, Ohio, is believed to be the oldest survivor of progeria at 44 years old as of September 2022.[81]
Alexandra Peraut is a Catalan girl with progeria; she has inspired the book Una nena entre vint milions ('A girl in 20 million'), a children's book to explain progeria to youngsters.[82][83]
Adalia Rose Williams, born December 10, 2006, was an American girl with progeria, who was a notable YouTuber and vlogger who shared her everyday life on social media. She died on January 12, 2022, at the age of 15.[84]
References
- James W, Berger T, Elston D (2005). Andrews' Diseases of the Skin: Clinical Dermatology (10th ed.). Saunders. p. 574. ISBN 978-0-7216-2921-6.
- Rapini RP, Bolognia JL, Jorizzo JL (2007). Dermatology: 2-Volume Set. St. Louis: Mosby. ISBN 978-1-4160-2999-1.
- Dictionary Reference: Progeria Archived 2013-04-10 at the Wayback Machine
- The Free Dictionary: Progeria Archived 2013-05-15 at the Wayback Machine
- "Hutchinson–Gilford Progeria – NORD (National Organization for Rare Disorders)". NORD (National Organization for Rare Disorders). 2014. Archived from the original on April 13, 2020. Retrieved 21 April 2017.
- "FDA Approves First Treatment for Hutchinson-Gilford Progeria Syndrome and Some Progeroid Laminopathies". U.S. Food and Drug Administration (FDA) (Press release). 20 November 2020. Retrieved 20 November 2020.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - "Drug Trials Snapshots: Zokinvy". U.S. Food and Drug Administration. 20 November 2020. Retrieved 11 December 2020. This article incorporates text from this source, which is in the public domain.
- Roach ES, Miller VS (2004). Neurocutaneous Disorders. Cambridge University Press. p. 150. ISBN 978-0-521-78153-4.
- Hsiao KJ (1998). Advances in Clinical Chemistry (33rd ed.). Academic Press. p. 10. ISBN 978-0-12-010333-1.
- Olive M, Harten I, Mitchell R, Beers JK, Djabali K, Cao K, et al. (November 2010). "Cardiovascular pathology in Hutchinson-Gilford progeria: correlation with the vascular pathology of aging". Arteriosclerosis, Thrombosis, and Vascular Biology. 30 (11): 2301–2309. doi:10.1161/ATVBAHA.110.209460. PMC 2965471. PMID 20798379.
- Sinha JK, Ghosh S, Raghunath M (May 2014). "Progeria: a rare genetic premature ageing disorder". The Indian Journal of Medical Research. 139 (5): 667–674. PMC 4140030. PMID 25027075.
- Hall H (8 December 2020). "Progeria". Science-Based Medicine. Archived from the original on 23 April 2021. Retrieved 23 April 2021.
- Arancio W, Pizzolanti G, Genovese SI, Pitrone M, Giordano C (2014). "Epigenetic involvement in Hutchinson-Gilford progeria syndrome: a mini-review". Gerontology. 60 (3): 197–203. doi:10.1159/000357206. hdl:10447/93705. PMID 24603298. S2CID 2459118.
- LMNA Archived 2015-12-28 at the Wayback Machine At Genes Archived 2015-12-22 at the Wayback Machine At Genetics Home Reference Archived 2019-02-04 at the Wayback Machine
- De Sandre-Giovannoli A, Bernard R, Cau P, Navarro C, Amiel J, Boccaccio I, et al. (June 2003). "Lamin a truncation in Hutchinson-Gilford progeria". Science. 300 (5628): 2055. doi:10.1126/science.1084125. PMID 12702809. S2CID 33927803.
- Cao K, Graziotto JJ, Blair CD, Mazzulli JR, Erdos MR, Krainc D, Collins FS (June 2011). "Rapamycin reverses cellular phenotypes and enhances mutant protein clearance in Hutchinson-Gilford progeria syndrome cells". Science Translational Medicine. 3 (89): 89ra58. doi:10.1126/scitranslmed.3002346. PMID 21715679. S2CID 206678031.
- Norris J (2011-10-21). "Aging Disease in Children Sheds Light on Normal Aging". UCSF web site. UCSF. Archived from the original on 2011-10-25. Retrieved 2011-10-25.
- Liu B, Wang J, Chan KM, Tjia WM, Deng W, Guan X, et al. (July 2005). "Genomic instability in laminopathy-based premature aging". Nature Medicine. 11 (7): 780–785. doi:10.1038/nm1266. PMID 15980864. S2CID 11798376.
- Chang W, Wang Y, Luxton GW, Östlund C, Worman HJ, Gundersen GG (February 2019). "Imbalanced nucleocytoskeletal connections create common polarity defects in progeria and physiological aging". Proceedings of the National Academy of Sciences of the United States of America. 116 (9): 3578–3583. Bibcode:2019PNAS..116.3578C. doi:10.1073/pnas.1809683116. PMC 6397528. PMID 30808750.
- "LMNA Gene" Archived 2017-10-12 at the Wayback Machine. GeneCards. Retrieved June 6, 2015.
- Zirn B, Kress W, Grimm T, Berthold LD, Neubauer B, Kuchelmeister K, et al. (April 2008). "Association of homozygous LMNA mutation R471C with new phenotype: mandibuloacral dysplasia, progeria, and rigid spine muscular dystrophy". American Journal of Medical Genetics. Part A. 146A (8): 1049–1054. doi:10.1002/ajmg.a.32259. PMID 18348272. S2CID 205309256.
- Cao H, Hegele RA (January 2000). "Nuclear lamin A/C R482Q mutation in canadian kindreds with Dunnigan-type familial partial lipodystrophy". Human Molecular Genetics. 9 (1): 109–112. doi:10.1093/hmg/9.1.109. PMID 10587585.
- Al-Haggar M, Madej-Pilarczyk A, Kozlowski L, Bujnicki JM, Yahia S, Abdel-Hadi D, et al. (November 2012). "A novel homozygous p.Arg527Leu LMNA mutation in two unrelated Egyptian families causes overlapping mandibuloacral dysplasia and progeria syndrome". European Journal of Human Genetics. 20 (11): 1134–1140. doi:10.1038/ejhg.2012.77. PMC 3476705. PMID 22549407.
- Agarwal AK, Kazachkova I, Ten S, Garg A (December 2008). "Severe mandibuloacral dysplasia-associated lipodystrophy and progeria in a young girl with a novel homozygous Arg527Cys LMNA mutation". The Journal of Clinical Endocrinology and Metabolism. 93 (12): 4617–4623. doi:10.1210/jc.2008-0123. PMC 2626450. PMID 18796515.
- Garg A, Cogulu O, Ozkinay F, Onay H, Agarwal AK (September 2005). "A novel homozygous Ala529Val LMNA mutation in Turkish patients with mandibuloacral dysplasia". The Journal of Clinical Endocrinology and Metabolism. 90 (9): 5259–5264. doi:10.1210/jc.2004-2560. PMID 15998779.
- Best BP (June 2009). "Nuclear DNA damage as a direct cause of aging". Rejuvenation Research. 12 (3): 199–208. CiteSeerX 10.1.1.318.738. doi:10.1089/rej.2009.0847. PMID 19594328.
- Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, et al. (May 2003). "Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome". Nature. 423 (6937): 293–298. Bibcode:2003Natur.423..293E. doi:10.1038/nature01629. hdl:2027.42/62684. PMID 12714972. S2CID 4420150.
- "Family Crisis Becomes Scientific Quest". Science. 300 (5621): 899. 9 May 2003. doi:10.1126/science.300.5621.899a. S2CID 220095842.
- Lans H, Hoeijmakers JH (March 2006). "Cell biology: ageing nucleus gets out of shape". Nature. 440 (7080): 32–34. Bibcode:2006Natur.440...32L. doi:10.1038/440032a. PMID 16511477. S2CID 4387289.
- Scaffidi P, Misteli T (May 2006). "Lamin A-dependent nuclear defects in human aging". Science. 312 (5776): 1059–1063. Bibcode:2006Sci...312.1059S. doi:10.1126/science.1127168. PMC 1855250. PMID 16645051.
- Haithcock E, Dayani Y, Neufeld E, Zahand AJ, Feinstein N, Mattout A, et al. (November 2005). "Age-related changes of nuclear architecture in Caenorhabditis elegans". Proceedings of the National Academy of Sciences of the United States of America. 102 (46): 16690–16695. Bibcode:2005PNAS..10216690H. doi:10.1073/pnas.0506955102. PMC 1283819. PMID 16269543.
- Zagorski N (November 2006). "A Comeback for the Ages: Lamin's connection with aging has reinvigorated research". Johns Hopkins University. Archived from the original on 2017-03-28. Retrieved 2020-05-22.
- Monterrubio-Ledezma F, Navarro-García F, Massieu L, Mondragón-Flores R, Soto-Ponce LA, Magaña JJ, Cisneros B (January 2023). "Rescue of Mitochondrial Function in Hutchinson-Gilford Progeria Syndrome by the Pharmacological Modulation of Exportin CRM1". Cells. 12 (2): 275. doi:10.3390/cells12020275. PMC 9856861. PMID 36672210.
- Xiong ZM, Choi JY, Wang K, Zhang H, Tariq Z, Wu D, et al. (April 2016). "Methylene blue alleviates nuclear and mitochondrial abnormalities in progeria". Aging Cell. 15 (2): 279–290. doi:10.1111/acel.12434. PMC 4783354. PMID 26663466.
- Feric M, Demarest TG, Tian J, Croteau DL, Bohr VA, Misteli T (March 2021). "Self-assembly of multi-component mitochondrial nucleoids via phase separation". The EMBO Journal. 40 (6): e107165. doi:10.15252/embj.2020107165. PMC 7957436. PMID 33619770.
- Maynard S, Hall A, Galanos P, Rizza S, Yamamoto T, Gram HH, et al. (September 2022). "Lamin A/C impairments cause mitochondrial dysfunction by attenuating PGC1α and the NAMPT-NAD+ pathway". Nucleic Acids Research. 50 (17): 9948–9965. doi:10.1093/nar/gkac741. PMC 9508839. PMID 36099415.
- "Learning About Progeria". genome.gov. Archived from the original on 16 April 2008. Retrieved 17 March 2008.
- "Progeria Research Foundation | The PRF Diagnostic Testing Program". Archived from the original on 28 August 2016. Retrieved 16 November 2011.
- "FDA Approves First Drug For Rare, Rapid-Aging Genetic Disorder". NPR.org. Archived from the original on 2020-11-22. Retrieved 2020-11-22.
- Gordon LB, Shappell H, Massaro J, D'Agostino RB, Brazier J, Campbell SE, et al. (April 2018). "Association of Lonafarnib Treatment vs No Treatment With Mortality Rate in Patients With Hutchinson-Gilford Progeria Syndrome". JAMA. 319 (16): 1687–1695. doi:10.1001/jama.2018.3264. PMC 5933395. PMID 29710166.
- "Progeria: Treatment". MayoClinic.com. Archived from the original on 2007-12-19. Retrieved 2008-03-17.
- Sadeghi-Nejad A, Demmer L (May 2007). "Growth hormone therapy in progeria". Journal of Pediatric Endocrinology & Metabolism. 20 (5): 633–637. doi:10.1515/jpem.2007.20.5.633. PMID 17642424. S2CID 5988572.
- Scaffidi P, Misteli T (April 2005). "Reversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndrome". Nature Medicine. 11 (4): 440–445. doi:10.1038/nm1204. PMC 1351119. PMID 15750600.
- Meta M, Yang SH, Bergo MO, Fong LG, Young SG (October 2006). "Protein farnesyltransferase inhibitors and progeria". Trends in Molecular Medicine. 12 (10): 480–487. doi:10.1016/j.molmed.2006.08.006. PMID 16942914.
- Clinical trial number NCT00425607 for "Phase II Trial of Lonafarnib (a Farnesyltransferase Inhibitor) for Progeria" at ClinicalTrials.gov
- "New Drug Hope for 'Aging' Kids". Science. 333 (6039): 142. 8 July 2011. doi:10.1126/science.333.6039.142-b.
- García-Aguirre I, Alamillo-Iniesta A, Rodríguez-Pérez R, Vélez-Aguilera G, Amaro-Encarnación E, Jiménez-Gutiérrez E, et al. (October 2019). "Enhanced nuclear protein export in premature aging and rescue of the progeria phenotype by modulation of CRM1 activity". Aging Cell. 18 (5): e13002. doi:10.1111/acel.13002. PMC 6718587. PMID 31305018.
- Ferreira BI, Cautain B, Grenho I, Link W (2020-05-07). "Small Molecule Inhibitors of CRM1". Frontiers in Pharmacology. 11: 625. doi:10.3389/fphar.2020.00625. PMC 7221118. PMID 32574233.
- Sternberg S (April 16, 2003). "Gene found for rapid aging disease in children". USA Today. Archived from the original on July 12, 2017. Retrieved December 12, 2013.
- "Progeria". MayoClinic.com. Archived from the original on May 11, 2008. Retrieved March 17, 2008.
- Brown WT (June 1992). "Progeria: a human-disease model of accelerated aging". The American Journal of Clinical Nutrition. 55 (6 Suppl): 1222S–1224S. doi:10.1093/ajcn/55.6.1222S. PMID 1590260.
- Corcoy R, Aris A, de Leiva A (June 1989). "Fertility in a case of progeria". The American Journal of the Medical Sciences. 297 (6): 383–384. doi:10.1097/00000441-198906000-00010. PMID 2735343. S2CID 72618179.
- Hennekam RC (December 2006). "Hutchinson-Gilford progeria syndrome: review of the phenotype". American Journal of Medical Genetics. Part A. 140 (23): 2603–2624. CiteSeerX 10.1.1.333.3746. doi:10.1002/ajmg.a.31346. PMID 16838330. S2CID 15692098.
- "Meet the Kids". Progeria Research Foundation. 1 September 2019. Archived from the original on 16 June 2019. Retrieved 29 September 2019.
- "Progeria Info". Archived from the original on 2 December 2013. Retrieved 28 November 2013.
- "In loving memory of those children who have passed away since The Progeria Research Foundation was formed in 1999". Progeria Research Foundation. 9 July 2019. Archived from the original on 16 June 2019. Retrieved 24 September 2019.
- "Progeria 101". Progeria Research Foundation. August 2019. Archived from the original on 16 June 2019. Retrieved 29 September 2019.
- "GLOBALHealthPR Co-Founder and Chair, John J. Seng, Receives Award from Progeria Research Foundation". Business Insider. 30 April 2018. Archived from the original on 20 April 2019. Retrieved 20 April 2019.
- Korf B (February 2008). "Hutchinson-Gilford progeria syndrome, aging, and the nuclear lamina". The New England Journal of Medicine. 358 (6): 552–555. doi:10.1056/NEJMp0800071. PMID 18256390. S2CID 44499453.
- Grant M (22 February 2005). "Family tormented by ageing disease". BBC News. Archived from the original on 19 July 2018.
- Koblan LW, Erdos MR, Wilson C, Cabral WA, Levy JM, Xiong ZM, et al. (January 2021). "In vivo base editing rescues Hutchinson-Gilford progeria syndrome in mice". Nature. 589 (7843): 608–614. Bibcode:2021Natur.589..608K. doi:10.1038/s41586-020-03086-7. PMC 7872200. PMID 33408413.
- LaMotte S (31 May 2022). "Changing our DNA: 'The age of human therapeutic gene editing is here'". CNN. Retrieved 2022-06-01.
- Zhang N, Hu Q, Sui T, Fu L, Zhang X, Wang Y, et al. (2023-02-02). "Unique progerin C-terminal peptide ameliorates Hutchinson–Gilford progeria syndrome phenotype by rescuing BUBR1". Nature Aging. 3 (2): 185–201. doi:10.1038/s43587-023-00361-w. ISSN 2662-8465. PMC 10154249. PMID 37118121. S2CID 256559744.
- Baker DJ, Jeganathan KB, Cameron JD, Thompson M, Juneja S, Kopecka A, et al. (July 2004). "BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice". Nature Genetics. 36 (7): 744–749. doi:10.1038/ng1382. PMID 15208629. S2CID 7871496.
- Redwood AB, Perkins SM, Vanderwaal RP, Feng Z, Biehl KJ, Gonzalez-Suarez I, et al. (August 2011). "A dual role for A-type lamins in DNA double-strand break repair". Cell Cycle. 10 (15): 2549–2560. doi:10.4161/cc.10.15.16531. PMC 3180193. PMID 21701264.
- Bernstein H, Payne CM, Bernstein C, Garewal H, Dvorak K (2008). "Cancer and aging as consequences of un-repaired DNA damage". In Kimura H, Suzuki A (eds.). New Research on DNA Damages. New York: Nova Science Publishers, Inc. pp. 1–47. ISBN 978-1-60456-581-2. Archived from the original on 2014-10-25.
- Horvath S, Oshima J, Martin GM, Lu AT, Quach A, Cohen H, et al. (July 2018). "Epigenetic clock for skin and blood cells applied to Hutchinson Gilford Progeria Syndrome and ex vivo studies". Aging. 10 (7): 1758–1775. doi:10.18632/aging.101508. PMC 6075434. PMID 30048243.
- "Royal Medical and Chirurgical Society". The Lancet. 127 (3272): 922–923. May 1886. doi:10.1016/S0140-6736(02)06582-0. p. 923:
Mr. JONATHAN HUTCHINSON contributed a paper on a case of congenital Absence of Hair, with Atrophic Condition of the Skin and its Appendages, in a boy whose mother had been almost wholly bald from alopecia areata from the age of six. This paper described the case of a boy three years and a half old, who presented a very withered 'old-manish' appearance.
- Kinmonth JB, Shepherd RC (November 1959). "Accidental injection of thiopentone into arteries: studies of pathology and treatment". British Medical Journal. 2 (5157): 914–918. doi:10.1136/bmj.2.5157.914. PMC 1990667. PMID 14409225.
- McClintock D, Ratner D, Lokuge M, Owens DM, Gordon LB, Collins FS, Djabali K (December 2007). "The mutant form of lamin A that causes Hutchinson-Gilford progeria is a biomarker of cellular aging in human skin". PLOS ONE. 2 (12): e1269. Bibcode:2007PLoSO...2.1269M. doi:10.1371/journal.pone.0001269. PMC 2092390. PMID 18060063.
- Merideth MA, Gordon LB, Clauss S, Sachdev V, Smith AC, Perry MB, et al. (February 2008). "Phenotype and course of Hutchinson-Gilford progeria syndrome". The New England Journal of Medicine. 358 (6): 592–604. doi:10.1056/NEJMoa0706898. PMC 2940940. PMID 18256394.
- "DICTIONARY". Archived from the original on 2016-03-04. Retrieved 2015-06-22.
- Confucius & Legge 2009, p. 113
- "I Am Not a Freak" (1987) at IMDb. Retrieved 2009-11-27.
- "The Aurora Encounter" (1986) at IMDb. Retrieved 2009-11-27.
- Patricia Montemurri (August 3, 2008). "'One of a kind': Little girl with progeria makes a big impact on loved ones". Detroit Free Press. Archived from the original on April 26, 2009.
- "Woman, Believe to be World's Oldest Progeriac, Dead At Age 29". The Associated Press. May 26, 1985. Archived from the original on November 5, 2019. Retrieved November 5, 2019.
- "My philosophy for a happy life - Sam Berns - TEDxMidAtlantic - YouTube". YouTube. Archived from the original on 2019-07-14. Retrieved 2016-11-26.
- "Hayley Okines, a teen trapped in a 104-year-old's body, dies at 17 - The Washington Post". The Washington Post. Archived from the original on 2019-12-04. Retrieved 2020-05-22.
- "Die Antwoord collaborator Leon Botha dies, age 26", Mail and Guardian, 6 June 2011, archived from the original on February 4, 2021, retrieved February 4, 2021
- Diaz A (12 September 2022). "One of the oldest living survivors of rapid aging disease at 44: 'Right now is all we have'". New York Post.
- 324cat (2021-02-28). ""Has vist mai una nena de 10 anys que és com una iaia?", el conte que explica la progèria". CCMA (in Catalan). Retrieved 2021-03-06.
- "Asociación Progeria ALEXANDRA PERAUT" (in Spanish). Retrieved 2021-07-28.
- King C (13 January 2022). "Texas YouTuber Adalia Rose dies at 15 after battle with rare genetic condition, family confirms". KSAT. Retrieved 13 January 2022.
Sources
- Legge J (2009). The Confucian Analects, the Great Learning & the Doctrine of the Mean. Cosimo, Inc. p. 113. ISBN 978-1-60520-643-1.