Thrombotic thrombocytopenic purpura
Thrombotic thrombocytopenic purpura (TTP) is a blood disorder that results in blood clots forming in small blood vessels throughout the body.[2] This results in a low platelet count, low red blood cells due to their breakdown, and often kidney, heart, and brain dysfunction.[1] Symptoms may include large bruises, fever, weakness, shortness of breath, confusion, and headache.[2][3] Repeated episodes may occur.[3]
| Thrombotic thrombocytopenic purpura | |
|---|---|
| Other names | Moschcowitz syndrome,[1] idiopathic thrombotic thrombocytopenic purpura[2] |
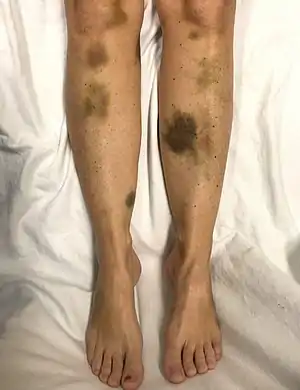 | |
| Spontaneous bruising in a woman with critically low platelets | |
| Specialty | Hematology |
| Symptoms | Large bruises, fever, weakness, shortness of breath, confusion, headache[3][2] |
| Usual onset | Adulthood[3] |
| Causes | Unknown, bacterial infections, certain medications, autoimmune diseases, pregnancy[3] |
| Diagnostic method | Based on symptoms and blood tests[2] |
| Differential diagnosis | Hemolytic-uremic syndrome (HUS), atypical hemolytic uremic syndrome (aHUS)[4] |
| Treatment | Plasma exchange, immunosuppressants[1] |
| Prognosis | < 20% risk of death[1] |
| Frequency | 1 in 100,000 people[3] |
In about half of cases a trigger is identified, while in the remainder the cause remains unknown.[3] Known triggers include bacterial infections, certain medications, autoimmune diseases such as lupus, and pregnancy.[3] The underlying mechanism typically involves antibodies inhibiting the enzyme ADAMTS13.[1] This results in decreased break down of large multimers of von Willebrand factor (vWF) into smaller units.[1] Less commonly TTP is inherited, known as Upshaw–Schulman syndrome, such that ADAMTS13 dysfunction is present from birth.[5] Diagnosis is typically based on symptoms and blood tests.[2] It may be supported by measuring activity of or antibodies against ADAMTS13.[2]
With plasma exchange the risk of death has decreased from more than 90% to less than 20%.[1] Immunosuppressants, such as glucocorticoids, and rituximab may also be used.[3] Platelet transfusions are generally not recommended.[6]
About 1 per 100,000 people are affected.[3] Onset is typically in adulthood and women are more often affected.[3] About 10% of cases begin in childhood.[3] The condition was first described by Eli Moschcowitz in 1924.[3] The underlying mechanism was determined in the 1980s and 1990s.[3]
Signs and symptoms
The signs and symptoms of TTP may at first be subtle and nonspecific. Many people experience an influenza-like or diarrheal illness before developing TTP.[7] Neurological symptoms are very common and vary greatly in severity. Frequently reported symptoms include feeling very tired, confusion, and headaches.[7] Seizures and symptoms similar to those of a stroke can also be seen.[7] Other symptoms include, but are not limited to jaundice or paleness of the skin, a fast heart rate or shortness of breath,[8] or dots on the skin known as petechiae.[9] High blood pressure has also been observed as a symptom.[10]
As TTP progresses, blood clots form within small blood vessels (microvasculature), and platelets (clotting cells) are consumed. As a result, bruising, and rarely bleeding can occur. The bruising often takes the form of purpura, while the most common site of bleeding, if it occurs, is from the nose or gums. Larger bruises (ecchymoses) may also develop.[11] The classic presentation of TTP, which occurs in less than 10% of people, includes five medical signs. These are:[3]
- Fever
- Changes in mental status
- Thrombocytopenia
- Reduced kidney function
- Hemolytic anemia (microangiopathic hemolytic anemia).[7]
Causes
TTP, as with other microangiopathic hemolytic anemias (MAHAs), is caused by spontaneous aggregation of platelets and activation of coagulation in the small blood vessels. Platelets are consumed in the aggregation process and bind vWF. These platelet-vWF complexes form small blood clots which circulate in the blood vessels and cause shearing of red blood cells, resulting in their rupture and formation of schistocytes.[12] The two best understood causes of TTP are due to autoimmunity (acquired TTP), caused by autoantibodies targeting ADAMTS13,[13] or congenital TTP: an inherited deficiency of ADAMTS13 (known as the Upshaw–Schulman syndrome).[12]
Autoimmune
In 1998, the majority of cases were shown to be caused by the inhibition of the enzyme ADAMTS13 by antibodies. Knowledge of this relationship between reduced ADAMTS13 and the pathogenesis of TTP is credited to two independent groups of researchers (Furlan and Tsai) who published their research in the same issue of the New England Journal of Medicine.[14]
ADAMTS13 is a metalloproteinase responsible for the breakdown of von Willebrand factor (vWF), a protein that links platelets, blood clots, and the blood vessel wall in the process of blood coagulation. Very large vWF multimers are more prone to lead to coagulation. Hence, without proper cleavage of vWF by ADAMTS13, coagulation occurs at a higher rate, especially in the microvasculature, part of the blood vessel system where vWF is most active due to high shear stress.[5]
Genetic

TTP may also be congenital. Such cases may be caused by mutations in the ADAMTS13 gene.[17][18] This hereditary form of TTP is called the Upshaw–Schulman syndrome (also spelled Upshaw–Schülman).[19][20] People with this inherited ADAMTS13 deficiency have a surprisingly mild phenotype, but develop TTP in clinical situations with increased von Willebrand factor levels, e.g. infection. Reportedly, less than 5% of all TTP cases are due to Upshaw–Schulman syndrome.[21] People with this syndrome generally have 5–10% of normal ADAMTS-13 activity.[22][23]
Secondary
Secondary TTP is diagnosed when the person's history mentions one of the known features associated with TTP. It comprises about 40% of all cases of TTP. Predisposing factors are:[12]
- Cancer
- Bone marrow transplantation
- Pregnancy
- Medication use:
- Antiviral drugs (acyclovir)
- Certain chemotherapy medications such as gemcitabine and mitomycin C
- Quinine
- Oxymorphone
- Quetiapine
- Bevacizumab
- Sunitinib
- Platelet aggregation inhibitors (ticlopidine, clopidogrel, and prasugrel)
- Immunosuppressants (ciclosporin, mitomycin, tacrolimus/FK506, interferon-α)
- Hormone altering drugs (estrogens, contraceptives, hormone replacement therapy)[24]
- HIV-1 infection
The mechanism of secondary TTP is poorly understood, as ADAMTS13 activity is generally not as depressed as in idiopathic TTP, and inhibitors cannot be detected. Probable etiology may involve, at least in some cases, endothelial damage,[25] although the formation of thrombi resulting in vessel occlusion may not be essential in the pathogenesis of secondary TTP.[26] These factors may also be considered a form of secondary aHUS; people presenting with these features are, therefore, potential candidates for anticomplement therapy.
Pathophysiology
The underlying mechanism typically involves autoantibody-mediated inhibition of the enzyme ADAMTS13, a metalloprotease responsible for cleaving large multimers of von Willebrand factor (vWF) into smaller units. The increase in circulating multimers of vWF increases platelet adhesion to areas of endothelial injury, particularly where arterioles and capillaries meet, which in turn results in the formation of small platelet clots called thrombi.[27] As platelets are used up in the formation of thrombi, this then leads to a decrease in the number of overall circulating platelets, which may then cause life-threatening bleeds. Red blood cells passing the microscopic clots are subjected to shear stress, which damages their membranes, leading to rupture of red blood cells within blood vessels,[9] which in turn leads to microangiopathic hemolytic anemia and schistocyte formation. The presence of the thrombi reduces blood flow to organs resulting in cellular injury and end organ damage.[13]
Recovery
Depression is common in those recovering from TTP; 59% of recovered TTP patients screened positive for depression within 11 years after recovery.[28]
Diagnosis
Differential diagnosis
TTP is a form of thrombotic microangiopathy (TMA),[29] the formation of blood clots in small blood vessels throughout the body, which can lead to microangiopathic hemolytic anemia and thrombocytopenia.[29] This characteristic is shared by two related syndromes, hemolytic-uremic syndrome (HUS) and atypical hemolytic uremic syndrome (aHUS).[4] Consequently, differential diagnosis of these TMA diseases is essential. Both TTP and HUS are characterized by fever, anemia, thrombocytopenia, renal failure, and neurological symptoms. Generally, TTP has higher rates of neurological symptoms (≤80%) and lower rates of renal symptoms (9%) than HUS (10–20% and 90%, respectively).[30]
Unlike HUS and aHUS,[31][32] TTP is known to be caused by a defect in the ADAMTS13 protein,[33] so a lab test showing ≤5% of normal ADAMTS13 levels is indicative of TTP.[27] ADAMTS13 levels above 5%, coupled with a positive test for shiga-toxin/enterohemorrhagic E. coli (EHEC), are more likely indicative of HUS,[34] whereas absence of shiga-toxin/EHEC can confirm a diagnosis of aHUS.[27]
Treatment
Due to the high mortality of untreated TTP, a presumptive diagnosis of TTP is made even when only microangiopathic hemolytic anemia and thrombocytopenia are seen, and therapy is started. Transfusion is contraindicated in thrombotic TTP, as it fuels the coagulopathy. Since the early 1990s, plasmapheresis has become the treatment of choice for TTP.[35][36] This is an exchange transfusion involving removal of the person's blood plasma through apheresis and replacement with donor plasma (fresh frozen plasma or cryosupernatant); the procedure must be repeated daily to eliminate the inhibitor and abate the symptoms. If apheresis is not available, fresh frozen plasma can be infused, but the volume that can be given safely is limited due to the danger of fluid overload.[37] Plasma infusion alone is not as beneficial as plasma exchange.[35] Corticosteroids (prednisone or prednisolone) are usually given.[36] Rituximab, a monoclonal antibody aimed at the CD20 molecule on B lymphocytes, may be used on diagnosis; this is thought to kill the B cells and thereby reduce the production of the inhibitor.[36] A stronger recommendation for rituximab exists where TTP does not respond to corticosteroids and plasmapheresis.[36]
Caplacizumab is an alternative option in treating TTP as it has been shown that it induces a faster disease resolution compared with those people who were on placebo.[38] However, the use of caplacizumab was associated with increase bleeding tendencies in some studied subjects.[39]
People with refractory or relapsing TTP may receive additional immunosuppressive therapy, e.g. vincristine, cyclophosphamide, cyclosporine A, or splenectomy.[3][37]
Children with Upshaw-Schulman syndrome receive prophylactic plasma every two to three weeks; this maintains adequate levels of functioning ADAMTS13. Some tolerate longer intervals between plasma infusions. Additional plasma infusions may be necessary for triggering events, such as surgery; alternatively, the platelet count may be monitored closely around these events with plasma being administered if the count drops.[40]
Measurements of blood levels of lactate dehydrogenase, platelets, and schistocytes are used to monitor disease progression or remission.[13][41] ADAMTS13 activity and inhibitor levels may be measured during follow-up, but in those without symptoms the use of rituximab is not recommended.[36]
Prognosis
The mortality rate is around 95% for untreated cases, but the prognosis is reasonably favorable (80–90% survival) for people with idiopathic TTP diagnosed and treated early with plasmapheresis.[42]
Epidemiology
The incidence of TTP is about 4–5 cases per million people per year.[43] Idiopathic TTP occurs more often in women as well as people of African descent, and TTP secondary to autoimmune disorders such as systemic lupus erythematosus occurs more frequently in people of African descent, although other secondary forms do not show this distribution.[44] Although Black people are at an increased risk for TTP, its presentation in Black people does not have any distinguishable features compared to those of other races.[45] Pregnant women and women in the post partum period accounted for a notable portion (12–31%) of the cases in some studies; TTP affects about one in 25,000 pregnancies.[46]
History
TTP was initially described by Eli Moschcowitz at the Beth Israel Hospital in New York City in 1924.[41][47] Moschcowitz ascribed the disease (incorrectly, as now known) to a toxic cause. Moschcowitz noted his patient, a 16-year-old girl, had anemia, small and large bruises, microscopic hematuria, and, at autopsy, disseminated microvascular thrombi.[48] In 1966, a review of 16 new cases and 255 previously reported cases led to the formulation of the classical pentad of symptoms and findings (i.e., thrombocytopenia, microangiopathic hemolytic anemia, neurological symptoms, kidney failure, fever); in this series, mortality rates were found to be very high (90%).[49]
While a response to blood transfusion had been noted before, a 1978 report and subsequent studies showed blood plasma was highly effective in improving the disease process.[50] In 1991, plasma exchange was reported to provide better response rates compared to plasma infusion.[51] In 1982, the disease had been linked with abnormally large von Willebrand factor multimers. The identification of a deficient protease in people with TTP was made in 1998. The location of ADAMTS13 within the human genome was identified in 2001.[50]
References
- Kremer Hovinga JA, Coppo P, Lämmle B, Moake JL, Miyata T, Vanhoorelbeke K (April 2017). "Thrombotic thrombocytopenic purpura". Nature Reviews Disease Primers. 3 17020. doi:10.1038/nrdp.2017.20. ISSN 2056-676X. PMID 28382967. S2CID 11960153.
- "Immune-mediated thrombotic thrombocytopenic purpura". Genetic and Rare Diseases Information Center. U.S. Department of Health & Human Services.
- Joly BS, Coppo P, Veyradier A (May 2017). "Thrombotic thrombocytopenic purpura". Blood. 129 (21): 2836–2846. doi:10.1182/blood-2016-10-709857. ISSN 0006-4971. PMID 28416507. S2CID 2543348.
- George JN (November 2010). "How I treat patients with thrombotic thrombocytopenic purpura: 2010". Blood. 116 (20): 4060–4069. doi:10.1182/blood-2010-07-271445. ISSN 0006-4971. PMID 20686117. S2CID 26844964.
- Moake JL (January 2004). "Von Willebrand factor, ADAMTS-13, and thrombotic thrombocytopenic purpura". Seminars in Hematology. 41 (1): 4–14. doi:10.1053/j.seminhematol.2003.10.003. ISSN 0037-1963. PMID 14727254.
- Wood, Marie E.; Philips, George K. (2003). Hematology/Oncology Secrets (3rd ed.). Elsevier. p. 68. ISBN 978-1-56053-516-4.
- Shatzel JJ, Taylor JA (March 2017). "Syndromes of Thrombotic Microangiopathy". Medical Clinics of North America. 101 (2): 395–415. doi:10.1016/j.mcna.2016.09.010. ISSN 0025-7125. PMID 28189178. S2CID 24123004.
- "Thrombotic thrombocytopenic purpura". MedlinePlus Genetics. National Library of Medicine.
- "Platelet Disorders - Thrombotic Thrombocytopenic Purpura (TTP)". National Heart, Lung, and Blood Institute.
- Shibagaki Y, Fujita T (January 2005). "Thrombotic Microangiopathy in Malignant Hypertension and Hemolytic Uremic Syndrome (HUS)/Thrombotic Thrombocytopenic Purpura (TTP): Can We Differentiate One from the Other?". Hypertension Research. 28 (1): 89–95. doi:10.1291/hypres.28.89. ISSN 1348-4214. PMID 15969259. S2CID 139098502.
- Chiasakul T, Cuker A (November 2018). "Clinical and laboratory diagnosis of TTP: an integrated approach". Hematology. 2018 (1): 530–538. doi:10.1182/asheducation-2018.1.530. ISSN 1520-4391. PMC 6246034. PMID 30504354. S2CID 54564230.
- Moake JL (August 2022). "Thrombotic Microangiopathies". New England Journal of Medicine. 347 (8): 589–600. doi:10.1056/NEJMra020528. ISSN 0028-4793. PMID 12192020. S2CID 40055198.
- Stanley M, Killeen RB, Michalski JM (January 2023). "Thrombotic Thrombocytopenic Purpura". StatPearls. StatPearls Publishing. PMID 28613472.
- Moake JL (November 1998). "Moschcowitz, Multimers, and Metalloprotease". New England Journal of Medicine. 339 (22): 1629–1631. doi:10.1056/NEJM199811263392210. ISSN 0028-4793. PMID 9828253.
- "THROMBOTIC THROMBOCYTOPENIC PURPURA, HEREDITARY; TTP". OMIM.
- "Thrombotic thrombocytopenic purpura". Orphanet.
- Conboy E, Partain PI, Warad D, Kluge ML, Arndt C, Chen D, Rodriguez V (January 2018). "A Severe Case of Congenital Thrombotic Thrombocytopenia Purpura Resulting From Compound Heterozygosity Involving a Novel ADAMTS13 Pathogenic Variant". Journal of Pediatric Hematology/Oncology. 40 (1): 60–62. doi:10.1097/MPH.0000000000000895. ISSN 1077-4114. PMID 28678087. S2CID 3470460.
- Lämmle B, Hovinga JA, George JN (February 2008). "Acquired thrombotic thrombocytopenic purpura: ADAMTS13 activity, anti-ADAMTS13 autoantibodies and risk of recurrent disease". Haematologica. 93 (2): 172–177. doi:10.3324/haematol.12701. ISSN 1592-8721. PMID 18245649. S2CID 17777111.
- Kremer Hovinga JA, George JN (October 2019). Longo DL (ed.). "Hereditary Thrombotic Thrombocytopenic Purpura". New England Journal of Medicine. 381 (17): 1653–1662. doi:10.1056/NEJMra1813013. ISSN 0028-4793. PMID 31644845. S2CID 204864996.
- Chapman K, Seldon M, Richards R (February 2012). "Thrombotic Microangiopathies, Thrombotic Thrombocytopenic Purpura, and ADAMTS-13". Seminars in Thrombosis and Hemostasis. 38 (1): 47–54. doi:10.1055/s-0031-1300951. ISSN 0094-6176. PMID 22314603. S2CID 13963783.
- Tsai H (2023). "Thrombotic Thrombocytopenic Purpura, Hemolytic-Uremic Syndrome, and Related Disorders". In Means RT Jr, Arber DA, Glader BE, Appelbaum FR, Rodgers GM, Dispenzieri A, Fehniger TA, Michaelis LC, Leonard JP (eds.). Wintrobe's Clinical Hematology. Vol. 2 (15th ed.). Wolters Kluwer. ISBN 978-1-975184-69-8.
- Levy GG, Nichols WC, Lian EC, Foroud T, McClintick JN, McGee BM, Yang AY, Siemieniak DR, Stark KR, Gruppo R, Sarode R, Shurin SB, Chandrasekaran V, Stabler SP, Sabio H (October 2001). "Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura". Nature. 413 (6855): 488–494. Bibcode:2001Natur.413..488L. doi:10.1038/35097008. hdl:2027.42/62592. ISSN 1476-4687. PMID 11586351. S2CID 4380010.
- Kokame K, Matsumoto M, Soejima K, Yagi H, Ishizashi H, Funato M, Tamai H, Konno M, Kamide K, Kawano Y, Miyata T, Fujimura Y (September 2002). "Mutations and common polymorphisms in ADAMTS13 gene responsible for von Willebrand factor-cleaving protease activity". Proceedings of the National Academy of Sciences. 99 (18): 11902–11907. Bibcode:2002PNAS...9911902K. doi:10.1073/pnas.172277399. ISSN 0027-8424. PMC 129366. PMID 12181489. S2CID 20496927.
- Menkes, John H.; Sarnat, Harvey B.; Maria, Bernard L. (2006). "Thrombotic Thrombocytopenic Purpura and Hemolytic-Uremic Syndrome". Child Neurology (7th ed.). Philadelphia: Lippincott Williams & Wilkins. p. 525. ISBN 9780781751049.
- van Mourik JA, Boertjes R, Huisveld IA, Fijnvandraat K, Pajkrt D, van Genderen PJ, Fijnheer R (1999-07-01). "von Willebrand Factor Propeptide in Vascular Disorders: A Tool to Distinguish Between Acute and Chronic Endothelial Cell Perturbation". Blood. 94 (1): 179–185. doi:10.1182/blood.V94.1.179.413k18_179_185. ISSN 1528-0020. PMID 10381511.
- Iwata H, Kami M, Hori A, Hamaki T, Takeuchi K, Mutou Y (June 2001). "An autopsy-based retrospective study of secondary thrombotic thrombocytopenic purpura". Haematologica. 86 (6): 669–70. PMID 11418383.
- Tsai H (January 2010). "Pathophysiology of thrombotic thrombocytopenic purpura". International Journal of Hematology. 91 (1): 1–19. doi:10.1007/s12185-009-0476-1. ISSN 1865-3774. PMC 3159000. PMID 20058209.
- Han B, Page EE, Stewart LM, Deford CC, Scott JG, Schwartz LH, Perdue JJ, Terrell DR, Vesely SK, George JN (August 2018). "Depression and cognitive impairment following recovery from thrombotic thrombocytopenic purpura: Depression and Cognitive Impairment Following TTP". American Journal of Hematology. 90 (8): 709–714. doi:10.1002/ajh.24060. PMC 4509840. PMID 25975932.
- Arnold DM, Patriquin CJ, Nazy I (January 2017). "Thrombotic microangiopathies: a general approach to diagnosis and management". CMAJ. 189 (4): E153–E159. doi:10.1503/cmaj.160142. ISSN 0820-3946. PMC 5266569. PMID 27754896.
- "MAHA, TTP, HUS, DIC... Oh My! Understanding Microangiopathic Hemolytic Anemias". Tampa Emergency Medicine Blog. University of South Florida. 2023-04-26. Retrieved 2023-08-23.
- Abrams CS (2012). "Thrombocytopenia". In Goldman L, Schafer AI (eds.). Goldman's Cecil Medicine. Vol. 1 (24th ed.). Elsevier. pp. 1124–1131. doi:10.1016/B978-1-4377-1604-7.00175-5. ISBN 978-1437716047.
In contrast to TTP, HUS is not caused by a deficiency of ADAMTS13.
- Feng S, Eyler SJ, Zhang Y, Maga T, Nester CM, Kroll MH, Smith RJ, Afshar-Kharghan V (August 2013). "Partial ADAMTS13 deficiency in atypical hemolytic uremic syndrome". Blood. 122 (8): 1487–1493. doi:10.1182/blood-2013-03-492421. ISSN 1528-0020. PMC 3750341. PMID 23847193.
Complement dysregulation leads to atypical hemolytic uremic syndrome (aHUS), while ADAMTS13 deficiency causes thrombotic thrombocytopenic purpura.
- Cataland, Spero R.; Wu, Haifeng M. (April 2014). "How I treat: the clinical differentiation and initial treatment of adult patients with atypical hemolytic uremic syndrome". Blood. 123 (16): 2478–2484. doi:10.1182/blood-2013-11-516237. ISSN 0006-4971. PMID 24599547.
- Bitzan M, Schaefer F, Reymond D (September 2010). "Treatment of typical (enteropathic) hemolytic uremic syndrome". Seminars in Thrombosis and Hemostasis. 36 (6): 594–610. doi:10.1055/s-0030-1262881. ISSN 1098-9064. PMID 20865636. S2CID 6129618.
- Michael, M; Elliott, EJ; Ridley, GF; Hodson, EM; Craig, JC (January 2009). "Interventions for haemolytic uraemic syndrome and thrombotic thrombocytopenic purpura". The Cochrane Database of Systematic Reviews. 1 (1): CD003595–CD003595–63. doi:10.1002/14651858.CD003595.pub2. hdl:10072/61440. PMC 7154575. PMID 19160220.
- Lim W, Vesely SK, George JN (March 2015). "The role of rituximab in the management of patients with acquired thrombotic thrombocytopenic purpura". Blood. 125 (10): 1526–31. doi:10.1182/blood-2014-10-559211. PMC 4351502. PMID 25573992.
- Allford SL, Hunt BJ, Rose P, Machin SJ (February 2003). "Guidelines on the diagnosis and management of the thrombotic microangiopathic haemolytic anaemias". British Journal of Haematology. 120 (4): 556–73. doi:10.1046/j.1365-2141.2003.04049.x. PMID 12588343. S2CID 38774829.
- Peyvandi F, Scully M, Kremer Hovinga JA, Cataland S, Knöbl P, Wu H, Artoni A, Westwood J, Mansouri Taleghani M (February 2016). "Caplacizumab for Acquired Thrombotic Thrombocytopenic Purpura". New England Journal of Medicine. 374 (6): 511–522. doi:10.1056/NEJMoa1505533. hdl:2434/829303. ISSN 0028-4793. PMID 26863353. S2CID 19482364.
- Goshua G, Bendapudi PK (December 2022). "Evidence-Based Minireview: Should caplacizumab be used routinely in unselected patients with immune thrombotic thrombocytopenic purpura?". Hematology. 2022 (1): 491–494. doi:10.1182/hematology.2022000412. ISSN 1520-4391. PMC 9820987. PMID 36485149.
- Loirat C, Girma J, Desconclois C, Coppo P, Veyradier A (January 2009). "Thrombotic thrombocytopenic purpura related to severe ADAMTS13 deficiency in children". Pediatric Nephrology. 24 (1): 19–29. doi:10.1007/s00467-008-0863-5. ISSN 1432-198X. PMID 18574602. S2CID 22209831.
- Saha M, McDaniel J, Zheng X (October 2010). "Thrombotic thrombocytopenic purpura: pathogenesis, diagnosis and potential novel therapeutics". Journal of Thrombosis and Haemostasis. 15 (10): 1889–1900. doi:10.1111/jth.13764. ISSN 1538-7836. PMC 5630501. PMID 28662310. S2CID 40493167.
- Tsai H (February 2006). "Current Concepts in Thrombotic Thrombocytopenic Purpura". Annual Review of Medicine. 57: 419–436. doi:10.1146/annurev.med.57.061804.084505. PMC 2426955. PMID 16409158.
- Terrell DR, Williams LA, Vesely SK, Lämmle B, Hovinga JA, George JN (July 2005). "The incidence of thrombotic thrombocytopenic purpura-hemolytic uremic syndrome: all patients, idiopathic patients, and patients with severe ADAMTS-13 deficiency". Journal of Thrombosis and Haemostasis. 3 (7): 1432–6. doi:10.1111/j.1538-7836.2005.01436.x. PMID 15978100. S2CID 24914279.
- Terrell DR, Vesely SK, Kremer Hovinga JA, Lämmle B, George JN (November 2010). "Different disparities of gender and race among the thrombotic thrombocytopenic purpura and hemolytic-uremic syndromes". American Journal of Hematology. 85 (11): 844–7. doi:10.1002/ajh.21833. PMC 3420337. PMID 20799358. S2CID 23118040.
- Martino, Suella; Jamme, Mathieu; Deligny, Christophe; Busson, Marc; Loiseau, Pascale; Azoulay, Elie; Galicier, Lionel; Pène, Frédéric; Provôt, François; Dossier, Antoine; Saheb, Samir; Veyradier, Agnès; Coppo, Paul; Microangiopathies, French Reference Center for Thrombotic (2016-07-06). "Thrombotic Thrombocytopenic Purpura in Black People: Impact of Ethnicity on Survival and Genetic Risk Factors". PLOS ONE. 11 (7): e0156679. Bibcode:2016PLoSO..1156679M. doi:10.1371/journal.pone.0156679. ISSN 1932-6203. PMC 4934773. PMID 27383202.
- Zheng XL, Sadler JE (2008). "Pathogenesis of Thrombotic Microangiopathies". Annual Review of Pathology. 3: 249–277. doi:10.1146/annurev.pathmechdis.3.121806.154311. PMC 2582586. PMID 18215115.
- Sukumar S, Lämmle B, Cataland SR (January 2021). "Thrombotic Thrombocytopenic Purpura: Pathophysiology, Diagnosis, and Management". Journal of Clinical Medicine. 10 (3): 536. doi:10.3390/jcm10030536. ISSN 2077-0383. PMC 7867179. PMID 33540569.
- Moschcowitz E (1924). "Hyaline Thrombosis of the Terminal Arterioles and Capillaries: A Hitherto Undescribed Disease". Proceedings of the New York Pathological Society. 24: 21–24.
- Amorosi EL, Ultmann JE (March 1966). "Thrombotic thrombocytopenic purpura: report of 16 cases and review of the literature". Medicine. 45 (2): 139. doi:10.1097/00005792-196603000-00003. S2CID 71943329.
- Sadler, JE (2008). "Von Willerbrand factor, ADAMTS13, and thrombotic thrombocytopenic purpura". Blood. 112 (1): 11–18. doi:10.1182/blood-2008-02-078170. PMC 2435681. PMID 18574040.
- Rock GA, Shumak KH, Buskard NA, et al. (August 1991). "Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. Canadian Apheresis Study Group". New England Journal of Medicine. 325 (6): 393–7. doi:10.1056/NEJM199108083250604. PMID 2062330.