Radical cyclization
Radical cyclization reactions are organic chemical transformations that yield cyclic products through radical intermediates. They usually proceed in three basic steps: selective radical generation, radical cyclization, and conversion of the cyclized radical to product.[1]
Introduction
Radical cyclization reactions produce mono- or polycyclic products through the action of radical intermediates. Because they are intramolecular transformations, they are often very rapid and selective. Selective radical generation can be achieved at carbons bound to a variety of functional groups, and reagents used to effect radical generation are numerous. The radical cyclization step usually involves the attack of a radical on a multiple bond. After this step occurs, the resulting cyclized radicals are quenched through the action of a radical scavenger, a fragmentation process, or an electron-transfer reaction. Five- and six-membered rings are the most common products; formation of smaller and larger rings is rarely observed.
Three conditions must be met for an efficient radical cyclization to take place:
- A method must be available to generate a radical selectively on the substrate.
- Radical cyclization must be faster than trapping of the initially formed radical.[2]
- All steps must be faster than undesired side reactions such as radical recombination or reaction with solvent.
Advantages: because radical intermediates are not charged species, reaction conditions are often mild and functional group tolerance is high and orthogonal to that of many polar processes. Reactions can be carried out in a variety of solvents (including arenes, alcohols, and water), as long as the solvent does not have a weak bond that can undergo abstraction, and products are often synthetically useful compounds that can be carried on using existing functionality or groups introduced during radical trapping.
Disadvantages: the relative rates of the various stages of radical cyclization reactions (and any side reactions) must be carefully controlled so that cyclization and trapping of the cyclized radical is favored. Side reactions are sometimes a problem, and cyclization is especially slow for small and large rings (although macrocyclizations, which resemble intermolecular radical reactions, are often high yielding).
Mechanism and stereochemistry
Prevailing mechanism
Because many reagents exist for radical generation and trapping, establishing a single prevailing mechanism is not possible. However, once a radical is generated, it can react with multiple bonds in an intramolecular fashion to yield cyclized radical intermediates. The two ends of the multiple bond constitute two possible sites of reaction. If the radical in the resulting intermediate ends up outside of the ring, the attack is termed "exo"; if it ends up inside the newly formed ring, the attack is called "endo." In many cases, exo cyclization is favored over endo cyclization (macrocyclizations constitute the major exception to this rule). 5-hexenyl radicals are the most synthetically useful intermediates for radical cyclizations, because cyclization is extremely rapid and exo selective.[3] Although the exo radical is less thermodynamically stable than the endo radical, the more rapid exo cyclization is rationalized by better orbital overlap in the chair-like exo transition state (see below).
(1)
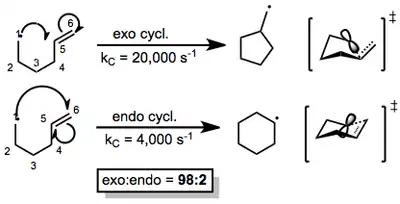
Substituents that affect the stability of these transition states can have a profound effect on the site selectivity of the reaction. Carbonyl substituents at the 2-position, for instance, encourage 6-endo ring closure. Alkyl substituents at positions 2, 3, 4, or 6 enhance selectivity for 5-exo closure.
Cyclization of the homologous 6-heptenyl radical is still selective, but is much slower—as a result, competitive side reactions are an important problem when these intermediates are involved. Additionally, 1,5-shifts can yield stabilized allylic radicals at comparable rates in these systems. In 6-hexenyl radical substrates, polarization of the reactive double bond with electron-withdrawing functional groups is often necessary to achieve high yields.[4] Stabilizing the initially formed radical with electron-withdrawing groups provides access to more stable 6-endo cyclization products preferentially.
(2)
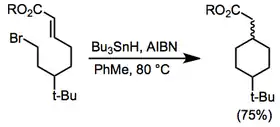
Cyclization reactions of vinyl, aryl, and acyl radicals are also known. Under conditions of kinetic control, 5-exo cyclization takes place preferentially. However, low concentrations of a radical scavenger establish thermodynamic control and provide access to 6-endo products—not via 6-endo cyclization, but by 5-exo cyclization followed by 3-exo closure and subsequent fragmentation (Dowd-Beckwith rearrangement). Whereas at high concentrations of the exo product is rapidly trapped preventing subsequent rearrangement to the endo product[5] Aryl radicals exhibit similar reactivity.
(3)
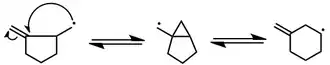
Cyclization can involve heteroatom-containing multiple bonds such as nitriles, oximes, and carbonyls. Attack at the carbon atom of the multiple bond is almost always observed.[6][7][8] In the latter case attack is reversible; however alkoxy radicals can be trapped using a stannane trapping agent.
Stereoselectivity
The diastereoselectivity of radical cyclizations is often high. In most all-carbon cases, selectivity can be rationalized according to Beckwith's guidelines, which invoke the reactant-like, exo transition state shown above.[9] Placing substituents in pseudoequatorial positions in the transition state leads to cis products from simple secondary radicals. Introducing polar substituents can favor trans products due to steric or electronic repulsion between the polar groups. In more complex systems, the development of transition state models requires consideration of factors such as allylic strain and boat-like transition states[10]
(4)
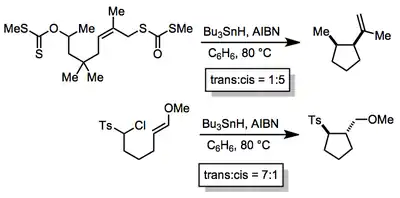
Chiral auxiliaries have been used in enantioselective radical cyclizations with limited success.[11] Small energy differences between early transition states constitute a profound barrier to success in this arena. In the example shown, diastereoselectivity (for both configurations of the left-hand stereocenter) is low and enantioselectivity is only moderate.
(5)
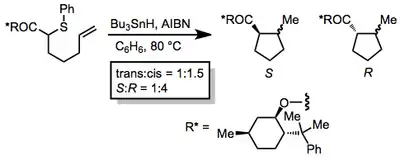
Substrates with stereocenters between the radical and multiple bond are often highly stereoselective. Radical cyclizations to form polycyclic products often take advantage of this property.[12]
Scope and limitations
Radical generation methods
The use of metal hydrides (tin, silicon and mercury hydrides) is common in radical cyclization reactions; the primary limitation of this method is the possibility of reduction of the initially formed radical by H-M. Fragmentation methods avoid this problem by incorporating the chain-transfer reagent into the substrate itself—the active chain-carrying radical is not released until after cyclization has taken place. The products of fragmentation methods retain a double bond as a result, and extra synthetic steps are usually required to incorporate the chain-carrying group.
Atom-transfer methods rely on the movement of an atom from the acyclic starting material to the cyclic radical to generate the product.[13][14] These methods use catalytic amounts of weak reagents, preventing problems associated with the presence of strong reducing agents (such as tin hydride). Hydrogen- and halogen-transfer processes are known; the latter tend to be more synthetically useful.
(6)
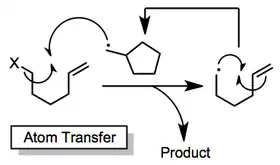
Oxidative[15] and reductive[16] cyclization methods also exist. These procedures require fairly electrophilic and nucleophilic radicals, respectively, to proceed effectively. Cyclic radicals are either oxidized or reduced and quenched with either external or internal nucleophiles or electrophiles, respectively.
Ring sizes
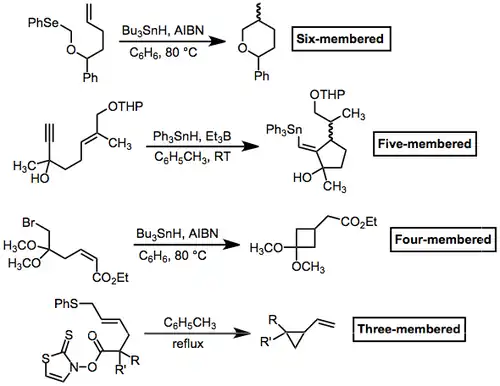
In general, radical cyclization to produce small rings is difficult. However, it is possible to trap the cyclized radical before re-opening. This process can be facilitated by fragmentation (see the three-membered case below) or by stabilization of the cyclized radical (see the four-membered case). Five- and six-membered rings are the most common sizes produced by radical cyclization.
(7)
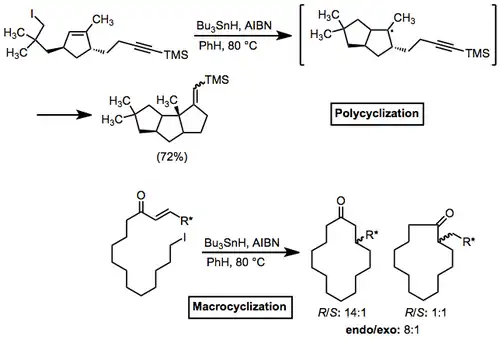
Polycycles and macrocycles can also be formed using radical cyclization reactions. In the former case, rings can be pre-formed and a single ring closed with radical cyclization, or multiple rings can be formed in a tandem process (as below).[17] Macrocyclizations, which lack the FMO requirement of cyclizations of smaller substrates, have the unique property of exhibiting endo selectivity.
(8)
Comparison with other methods
In comparison to cationic cyclizations, radical cyclizations avoid issues associated with Wagner-Meerwein rearrangements, do not require strongly acidic conditions, and can be kinetically controlled. Cationic cyclizations are usually thermodynamically controlled. Radical cyclizations are much faster than analogous anionic cyclizations, and avoid β-elimination side reactions. Anionic Michael-type cyclization is an alternative to radical cyclization of activated olefins. Metal-catalyzed cyclization reactions usually require mildly basic conditions, and substrates must be chosen to avoid β-hydride elimination. The primary limitation of radical cyclizations with respect to these other methods is the potential for radical side reactions.
Experimental conditions and procedure
Typical conditions
Radical reactions must be carried out under inert atmosphere as dioxygen is a triplet radical which will intercept radical intermediates. Because the relative rates of a number of processes are important to the reaction, concentrations must be carefully adjusted to optimize reaction conditions. Reactions are generally carried out in solvents whose bonds have high bond dissociation energies (BDEs), including benzene, methanol or benzotrifluoride. Even aqueous conditions are tolerated,[18] since water has a strong O-H bond with a BDE of 494 kJ/mol. This is in contrast to many polar processes, where hydroxylic solvents (or polar X-H bonds in the substrate itself) may not be tolerated due to the nucleophilicity or acidity of the functional group.
Example procedure
(9)
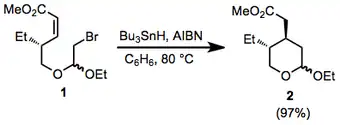
A mixture of bromo acetal 1 (549 mg, 1.78 mmol), AIBN (30.3 mg, 0.185 mmol), and Bu3SnH (0.65 mL, 2.42 mmol) in dry benzene (12 mL) was heated under reflux for 1 hour and then evaporated under reduced pressure. Silicagel column chromatography of the crude product with hexane–EtOAc (92:8) as eluant gave tetrahydropyran 2 (395 mg, 97%) as an oily mixture of two diastereomers. (c 0.43, CHCl3); IR (CHCl3):1732 cm–1;1H NMR (CDCl3)δ 4.77–4.89 (m, 0.6H), 4.66–4.69 (m, 0.4H), 3.40–4.44 (m, 4H), 3.68 (s, 3H), 2.61 (dd, J = 15.2, 4.2 Hz, 1H), 2.51 (dd, J = 15.2, 3.8 Hz, 1H), 0.73–1.06 (m, 3H); mass spectrum: m/z 215 (M+–Me); Anal. Calcd for C12H22O4: C, 62.6; H, 9.65. Found: C, 62.6; H, 9.7.[19]
References
- Giese, B.; Kopping, B.; Gobel, T.; Dickhaut, J.; Thoma, G.; Kulicke, K. J.; Trach., F. Org. React. 1996, 48, 301-361. doi:10.1002/0471264180.or048.02
- A lower limit on the rate of the cyclization step is 100 s−1.
- Beckwith, A.; Schiesser, C. Tetrahedron 1985, 41, 3925.
- Hanessian, S.; Dhanoa, D.; Beaulieu, P. Can. J. Chem. 1987, 65, 1859.
- Beckwith, A.; O'Shea, D. Tetrahedron Lett. 1986, 27, 4525.
- Tsang, R.; Dickson, J.; Pak, H.; Walton, R.; Fraser-Reid, B. J. Am. Chem. Soc. 1987, 104, 3484.
- Bartlett, P.; McLaren, K.; Ting, P. J. Am. Chem. Soc. 1988, 110, 1633.
- Clive, D.; Beaulieu, P.; Set, L. J. Org. Chem. 1984, 49, 1313.
- Beckwith, A.; Christopher, J.; Lawrence, T.; Serelis, A. Aust. J. Chem. 1983, 36, 545.
- RajanBabu, T. V. Acc. Chem. Res. 1991, 24, 139.
- Chen, M.-Y.; Fang, J.-M.; Tsai, Y.-M.; Yeh, R.-L. J. Chem. Soc., Chem. Commun., 1991, 1603.
- Stork, G.; Sher, P. M.; Chen, H. L. J. Am. Chem. Soc. 1986, 108, 6384.
- Julia, M.; Maumy, M. Org. Synth. 1976, 55, 57.
- Iqbal, J.; Bhatia, B.; Nayyar, N. Chem. Rev. 1994, 94, 519.
- Corey, E.; Kang, M. J. Am. Chem. Soc. 1984, 106, 5384.
- Nugent, W.; RajanBabu, T. J. Am. Chem. Soc. 1988, 110, 8561.
- Curran, Dennis P.; Rakiewicz, Donna M. (1985). "Tandem radical approach to linear condensed cyclopentanoids. Total synthesis of (.+-.)-hirsutene". Journal of the American Chemical Society. 107 (5): 1448–1449. doi:10.1021/ja00291a077.
- Yorimitsu, Hideki; Nakamura, Tomoaki; Shinokubo, Hiroshi; Oshima, Koichiro; Omoto, Kiyoyuki; Fujimoto, Hiroshi (November 2000). "Powerful Solvent Effect of Water in Radical Reaction: Triethylborane-Induced Atom-Transfer Radical Cyclization in Water". Journal of the American Chemical Society. 122 (45): 11041–11047. doi:10.1021/ja0014281. ISSN 0002-7863.
- Ikara, M.; Yasai, K.; Tanigachi, N.; Fukumoto, K. J. Chem. Soc., Perkin Trans. 1, 1990, 1469.