Thermodynamic versus kinetic reaction control
Thermodynamic reaction control or kinetic reaction control in a chemical reaction can decide the composition in a reaction product mixture when competing pathways lead to different products and the reaction conditions influence the selectivity or stereoselectivity. The distinction is relevant when product A forms faster than product B because the activation energy for product A is lower than that for product B, yet product B is more stable. In such a case A is the kinetic product and is favoured under kinetic control and B is the thermodynamic product and is favoured under thermodynamic control.[1][2][3]
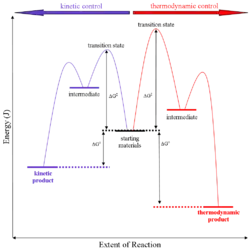
The conditions of the reaction, such as temperature, pressure, or solvent, affect which reaction pathway may be favored: either the kinetically controlled or the thermodynamically controlled one. Note this is only true if the activation energy of the two pathways differ, with one pathway having a lower Ea (energy of activation) than the other.
Prevalence of thermodynamic or kinetic control determines the final composition of the product when these competing reaction pathways lead to different products. The reaction conditions as mentioned above influence the selectivity of the reaction - i.e., which pathway is taken.
Asymmetric synthesis is a field in which the distinction between kinetic and thermodynamic control is especially important. Because pairs of enantiomers have, for all intents and purposes, the same Gibbs free energy, thermodynamic control will produce a racemic mixture by necessity. Thus, any catalytic reaction that provides product with nonzero enantiomeric excess is under at least partial kinetic control. (In many stoichiometric asymmetric transformations, the enantiomeric products are actually formed as a complex with the chirality source before the workup stage of the reaction, technically making the reaction a diastereoselective one. Although such reactions are still usually kinetically controlled, thermodynamic control is at least possible, in principle.)
Scope
In Diels–Alder reactions
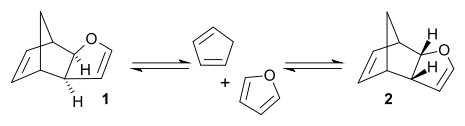
The Diels–Alder reaction of cyclopentadiene with furan can produce two isomeric products. At room temperature, kinetic reaction control prevails and the less stable endo isomer 2 is the main reaction product. At 81 °C and after long reaction times, the chemical equilibrium can assert itself and the thermodynamically more stable exo isomer 1 is formed.[4] The exo product is more stable by virtue of a lower degree of steric congestion, while the endo product is favoured by orbital overlap in the transition state.
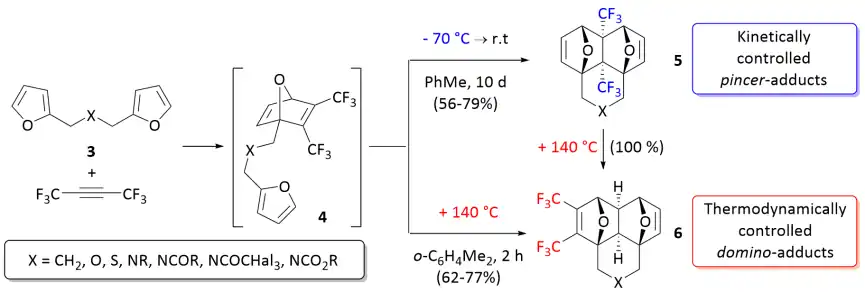
An outstanding and very rare example of the full kinetic and thermodynamic reaction control in the process of the tandem inter-/intramolecular Diels–Alder reaction of bis-furyl dienes 3 with hexafluoro-2-butyne or dimethyl acetylenedicarboxylate (DMAD) have been discovered and described in 2018.[5][6] At low temperature, the reactions occur chemoselectively leading exclusively to adducts of pincer-[4+2] cycloaddition (5). The exclusive formation of domino-adducts (6) is observed at elevated temperatures.
Theoretical DFT calculations of the reaction between hexafluoro-2-butyne and dienes 3a-c were performed. The reaction starting with [4+2] cycloaddition of CF3C≡CCF3 at one of the furan moieties occurs in a concerted fashion via TS1 and represents the rate limiting step of the whole process with the activation barrier ΔG‡ ≈ 23.1–26.8 kcal/mol.
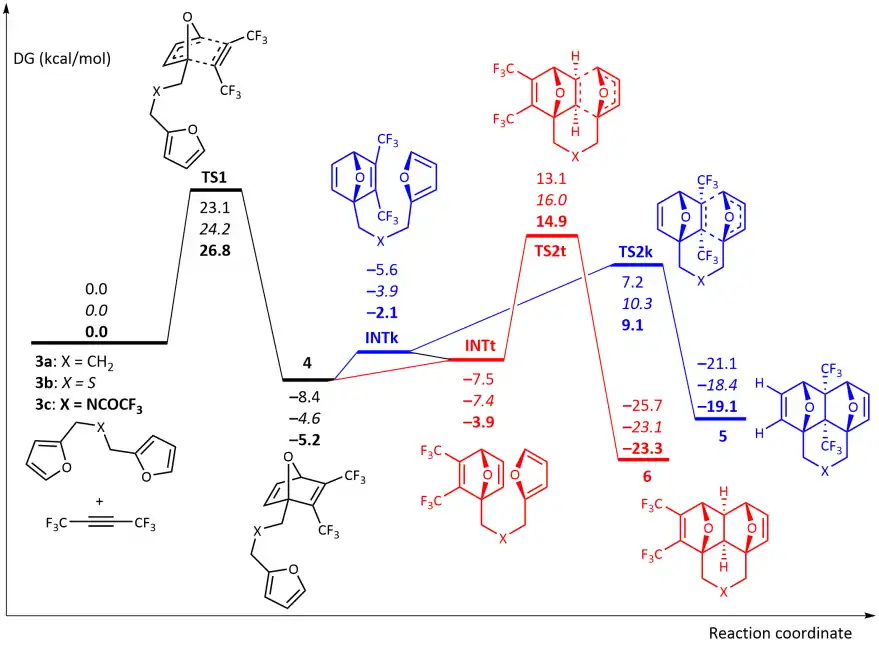
Further, the reaction could proceed via two competing channels, i.e. either leading to the pincer type products 5 via TS2k or resulting in the formation of the domino product 6 via TS2t. The calculations showed that the first channel is more kinetically favourable (ΔG‡ ≈ 5.7–5.9 kcal/mol). Meanwhile, the domino products 6 are more thermodynamically stable than 5 (ΔG‡ ≈ 4.2-4.7 kcal/mol) and this fact may cause isomerization of 5 into 6 at elevated temperature. Indeed, the calculated activation barriers for the 5 → 6 isomerization via the retro-Diels–Alder reaction of 5 followed by the intramolecular [4+2]-cycloaddition in the chain intermediate 4 to give 6 are 34.0–34.4 kcal/mol.
In enolate chemistry
In the protonation of an enolate ion, the kinetic product is the enol and the thermodynamic product is a ketone or aldehyde. Carbonyl compounds and their enols interchange rapidly by proton transfers catalyzed by acids or bases, even in trace amounts, in this case mediated by the enolate or the proton source.
In the deprotonation of an unsymmetrical ketone, the kinetic product is the enolate resulting from removal of the most accessible α-H while the thermodynamic product has the more highly substituted enolate moiety.[7][8][9][10] Use of low temperatures and sterically demanding bases increases the kinetic selectivity. Here, the difference in pKb between the base and the enolate is so large that the reaction is essentially irreversible, so the equilibration leading to the thermodynamic product is likely a proton exchange occurring during the addition between the kinetic enolate and as-yet-unreacted ketone. An inverse addition (adding ketone to the base) with rapid mixing would minimize this. The position of the equilibrium will depend on the countercation and solvent.
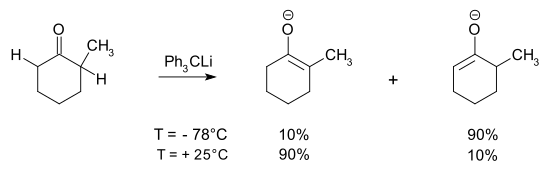
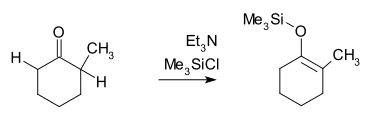
If a much weaker base is used, the deprotonation will be incomplete, and there will be an equilibrium between reactants and products. Thermodynamic control is obtained, however the reaction remains incomplete unless the product enolate is trapped, as in the example below. Since H transfers are very fast, the trapping reaction being slower, the ratio of trapped products largely mirrors the deprotonation equilibrium.
In electrophilic additions
The electrophilic addition reaction of hydrogen bromide to 1,3-butadiene above room temperature leads predominantly to the thermodynamically more stable 1,4 adduct, 1-bromo-2-butene, but decreasing the reaction temperature to below room temperature favours the kinetic 1,2 adduct, 3-bromo-1-butene.[3]
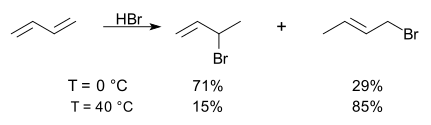
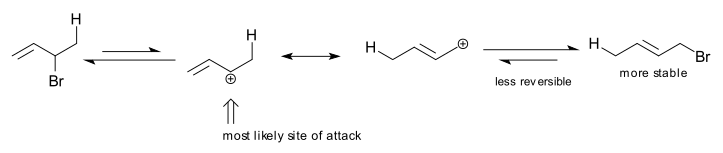
- The rationale for the differing selectivities is as follows: Both products result from Markovnikov protonation at position 1, resulting in a resonance-stabilized allylic cation. The 1,4 adduct places the larger Br atom at a less congested site and includes a more highly substituted alkene moiety, while the 1,2 adduct is the result of the attack by the nucleophile (Br−) at the carbon of the allylic cation bearing the greatest positive charge (the more highly substituted carbon is the most likely place for the positive charge).
Characteristics
- In principle, every reaction is on the continuum between pure kinetic control and pure thermodynamic control. These terms are with respect to a given temperature and time scale. A process approaches pure kinetic control at low temperature and short reaction time. For a sufficiently long time scale, every reaction approaches pure thermodynamic control, at least in principle.[11][12] This time scale becomes shorter as the temperature is raised.
- In every reaction, the first product formed is that which is most easily formed. Thus, every reaction a priori starts under kinetic control.[13]
- A necessary condition for thermodynamic control is reversibility or a mechanism permitting the equilibration between products. Reactions are considered to take place under thermodynamic reaction control when the reverse reaction is sufficiently rapid that the equilibrium establishes itself within the allotted reaction time. In this way, the thermodynamically more stable product is always favoured.
- Under kinetic reaction control, one or both forward reactions leading to the possible products is significantly faster than the equilibration between the products. After reaction time t, the product ratio is the ratio of rate constants k and thus a function of the difference in activation energies Ea or ΔG‡:
- (equation 1)
![\ln \left({\frac {[A]_{t}}{[B]_{t}}}\right)=\ln \left({\frac {k_{A}}{k_{B}}}\right)=-{\frac {\Delta E_{a}}{RT}}](../I/d3c4abce8554f016a84af1e7dad83761b5dd71d7.svg)
- Unless equilibration is prevented (e.g., by removal of the product from the reaction mixture as soon as it forms), "pure" kinetic control is strictly speaking impossible, because some amount of equilibration will take place before the reactants are entirely consumed. In practice, many systems are well approximated as operating under kinetic control, due to negligibly slow equilibration. For example, many enantioselective catalytic systems provide nearly enantiopure product (> 99% ee), even though the enantiomeric products have the same Gibbs free energy and are equally favored thermodynamically.
- Under pure thermodynamic reaction control, when the equilibrium has been reached, the product distribution will be a function of the stabilities G°. After an infinite amount of reaction time, the ratio of product concentrations will equal the equilibrium constant Keq and therefore be a function of the difference in Gibbs free energies,
- (equation 2)
- In principle, "pure" thermodynamic control is also impossible, since equilibrium is only achieved after infinite reaction time. In practice, if A and B interconvert with overall rate constants kf and kr, then for most practical purposes, the change in composition becomes negligible after t ~ 3.5/(kf + kr), or approximately five half-lives, and the system product ratio can be regarded as the result of thermodynamic control.
![\ln \left({\frac {[A]_{{\infty }}}{[B]_{{\infty }}}}\right)=\ln \ K_{{eq}}=-{\frac {\Delta G^{\circ }}{RT}}](../I/fe18ae931f200f667234f9ff8c2033ba94c98205.svg)
- In general, short reaction times favour kinetic control, whereas longer reaction times favour thermodynamic reaction control. Low temperatures will enhance the selectivity under both sets of conditions, since T is in the denominator in both cases. The ideal temperature to optimise the yield of the fastest-forming product will be the lowest temperature that will ensure reaction completion in a reasonable amount of time.[14] The ideal temperature for a reaction under thermodynamic control is the lowest temperature at which equilibrium will be reached in a reasonable amount of time.[15] If needed, the selectivity can be increased by then slowly cooling the reaction mixture to shift the equilibrium further toward the most stable product. When the difference in product stability is very large, the thermodynamically controlled product can dominate even under relatively vigorous reaction conditions.
- If a reaction is under thermodynamic control at a given temperature, it will also be under thermodynamic control at a higher temperature for the same reaction time.
- In the same manner, if a reaction is under kinetic control at a given temperature, it will also be under kinetic control at any lower temperature for the same reaction time.
- If one presumes that a new reaction will be a priori under kinetic control, one can detect the presence of an equilibration mechanism (and therefore the possibility of thermodynamic control) if the product distribution:
- changes over time,
- shows one product to be dominant at one temperature while another dominates at a different temperature (inversion of dominance), or
- changes with temperature but is not consistent with equation 1, that is a change in temperature (without changing the reaction time) causes a change in the product ratio that is larger or smaller than would be expected from the change in temperature alone, assuming that is largely invariant with temperature over a modest temperature range.[16]
- In the same way, one can detect the possibility of kinetic control if a temperature change causes a change in the product ratio that is inconsistent with equation 2, assuming that is largely invariant with temperature over a modest temperature range.[17]
![{[A]_{t}}/{[B]_{t}}](../I/6e66460d660f8572d9b47c3943b17e49c35a4048.svg)


History
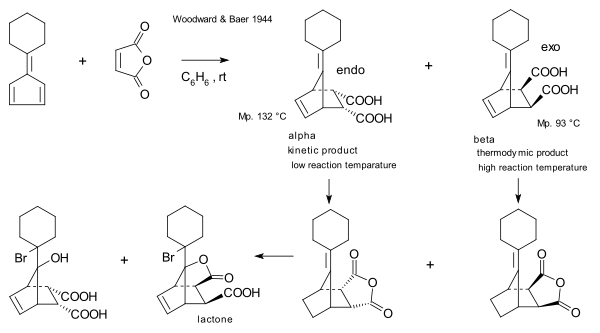
The first to report on the relationship between kinetic and thermodynamic control were R.B. Woodward and Harold Baer in 1944.[18] They were re-investigating a reaction between maleic anhydride and a fulvene first reported in 1929 by Otto Diels and Kurt Alder.[19] They observed that while the endo isomer is formed more rapidly, longer reaction times, as well as relatively elevated temperatures, result in higher exo / endo ratios which had to be considered in the light of the remarkable stability of the exo-compound on the one hand and the very facile dissociation of the endo isomer on the other.
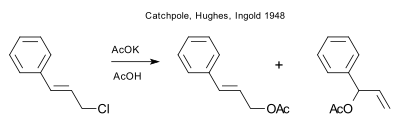
C. K. Ingold with E. D. Hughes and G. Catchpole independently described a thermodynamic and kinetic reaction control model in 1948.[20] They were reinvestigating a certain allylic rearrangement reported in 1930 by Jakob Meisenheimer.[21] Solvolysis of gamma-phenylallyl chloride with AcOK in acetic acid was found to give a mixture of the gamma and the alpha acetate with the latter converting to the first by equilibration. This was interpreted as a case in the field of anionotropy of the phenomenon, familiar in prototropy, of the distinction between kinetic and thermodynamic control in ion-recombination.
References
- Organic Chemistry, 3rd ed., M. A. Fox & J. K. Whitesell, Jones & Bartlett, 2004 ISBN 0-7637-2197-2
- A Guidebook to Mechanism in Organic Chemistry, 6th Edition, Peter Sykes, Pearson Prentice Hall, 1986. ISBN 0-582-44695-3
- Introduction to Organic Chemistry I, Seth Robert Elsheimer, Blackwell Publishing, 2000 ISBN 0-632-04417-9
- Advanced Organic Chemistry Part A: Structure and Mechanisms, 5th ed., Francis A. Carey, Richard J. Sundberg, 2007 ISBN 978-0-387-44899-2
- Kseniya K. Borisova, Elizaveta A. Kvyatkovskaya, Eugeniya V. Nikitina, Rinat R. Aysin, Roman A. Novikov, and Fedor I. Zubkov. “A Classical Example of Total Kinetic and Thermodynamic Control. The Diels-Alder Reaction between DMAD and Bis-furyl Dienes.” J. Org. Chem., 2018, 83 (8), pp 4840-4850. doi:10.1021/acs.joc.8b00336 https://pubs.acs.org/doi/abs/10.1021/acs.joc.8b00336
- Kseniya K. Borisova, Eugeniya V. Nikitina, Roman A. Novikov, Victor N. Khrustalev, Pavel V. Dorovatovskii, Yan V. Zubavichus, Maxim L. Kuznetsov, Vladimir P. Zaytsev, Alexey V. Varlamov and Fedor I. Zubkov. “Diels–Alder reactions between hexafluoro-2-butyne and bis-furyl dienes: kinetic versus thermodynamic control.” Chem. Commun., 2018, 54, pp 2850-2853. doi:10.1039/c7cc09466c http://pubs.rsc.org/en/content/articlelanding/2018/cc/c7cc09466c#!divAbstract
- Thermodynamic Product vs Kinetic Product
- Jean d'Angelo, Tetrahedron report number 25 : Ketone enolates: regiospecific preparation and synthetic uses, Tetrahedron, Volume 32, Issue 24, 1976, Pages 2979-2990, ISSN 0040-4020, doi:10.1016/0040-4020(76)80156-1
- The Chemistry of Carbanions. IX. The Potassium and Lithium Enolates Derived from Cyclic Ketones Herbert O. House, Barry M. Trost J. Org. Chem., 1965, 30 (5), pp 1341–1348 doi:10.1021/jo01016a001
- Chemistry of carbanions. XV. Stereochemistry of alkylation of 4-tert-butylcyclohexanone Herbert O. House, Ben A. Tefertiller, Hugh D. Olmstead J. Org. Chem., 1968, 33 (3), pp 935–942 doi:10.1021/jo01267a002
- Khopade, Tushar; Mete, Trimbak; Arora, Jyotsna; Bhat, Ramakrishna (2018). "An Adverse Effect of Higher Catalyst Loading and Longer Reaction Time on Enantioselectivity in an Organocatalytic Multicomponent Reaction". Chemistry: A European Journal. 24 (23): 6036–6040. doi:10.1002/chem.201800278. PMID 29465758.
- Rulli, Giuseppe; Duangdee, Nongnaphat; Baer, Katrin; Hummel, Werner; Berkessel, Albrecht; Gröger, Harald (2011). "Direction of Kinetically versus Thermodynamically Controlled Organocatalysis and Its Application in Chemoenzymatic Synthesis". Angewandte Chemie International Edition. 50 (34): 7944–7947. doi:10.1002/anie.201008042. PMID 21744441. S2CID 42971817.
- Only if a subsequent equilibration is as fast or faster is this not true.
- Unless one is content with an incomplete reaction, whence a separation of product from unreacted starting material may be necessary.
- At worst, Keq will approach 1 as T rises and the proportion of the most stable product will tend toward 50% of the reaction mixture.
- will be temperature-independent or nearly so if is small, which would be the case if the rate-determining steps leading to each product were of the same molecularity, for instance if both involved collisions with the same reactant.
- will be temperature-independent or nearly so if is small, which would be the case if the overall transformations to each product were of the same molecularity, for instance if both were fragmentations of a molecule to produce a pair of molecules or if both were condensations of two molecules to give a single molecule.
- Studies on Diene-addition Reactions. II.1 The Reaction of 6,6-Pentamethylenefulvene with Maleic Anhydride R. B. Woodward, Harold Baer J. Am. Chem. Soc., 1944, 66 (4), pp 645–649 doi:10.1021/ja01232a042
- Diels, O. and Alder, K. (1929), Synthesen in der hydroaromatischen Reihe, IV. Mitteilung: Über die Anlagerung von Maleinsäure-anhydrid an arylierte Diene, Triene und Fulvene (Mitbearbeitet von Paul Pries). Berichte der deutschen chemischen Gesellschaft (A and B Series), 62: 2081–2087. doi:10.1002/cber.19290620829
- Rearrangement and substitution in anionotropic systems. Part III. Mechanism of, and equilibrium in, anionotropic change A. G. Catchpole, E. D. Hughes and C. K. Ingold J. Chem. Soc., 1948, 8-17 doi:10.1039/JR9480000008
- Meisenheimer, J. and Link, J. (1930), Über die Verschiebung in der Allyl-Gruppe. 3. Mitteilung über Substitution und Addition. Justus Liebigs Annalen der Chemie, 479: 211–277. doi:10.1002/jlac.19304790114

