Reductions with metal alkoxyaluminium hydrides
Reductions with metal alkoxyaluminium hydrides are chemical reactions that involve either the net hydrogenation of an unsaturated compound or the replacement of a reducible functional group with hydrogen by metal alkoxyaluminium hydride reagents.[1][2]
Introduction
Sodium borohydride and lithium aluminium hydride are commonly used for the reduction of organic compounds.[3][4] These two reagents are on the extremes of reactivity—whereas lithium aluminium hydride reacts with nearly all reducible functional groups, sodium borohydride reacts with a much more limited range of functional groups. Diminished or enhanced reactivity may be realized by the replacement of one or more of the hydrogens in these reagents with alkoxy groups.
Additionally, substitution of hydrogen for chiral alkoxy groups in these reagents enables asymmetric reductions.[5] Although methods involving stoichiometric amounts of chiral metal hydrides have been supplanted in modern times by enantioselective catalytic reductions, they are of historical interest as early examples of stereoselective reactions.
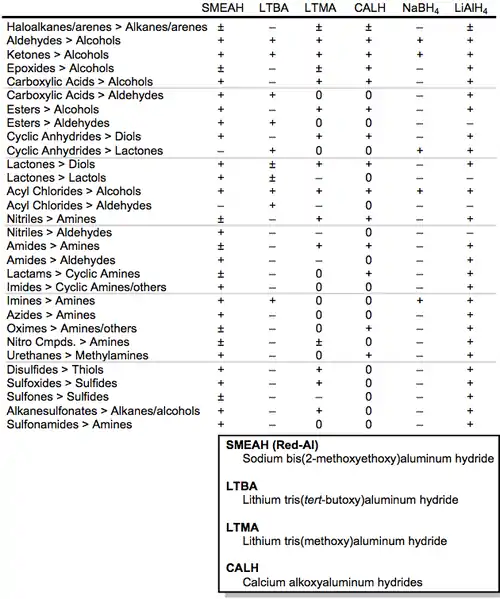
The table below summarizes the reductions that may be carried out with a variety of metal aluminium hydrides and borohydrides. The symbol "+" indicates that reduction does occur, "-" indicates that reduction does not occur, "±" indicates that reduction depends on the structure of the substrate, and "0" indicates a lack of literature information.
-
(1)
Mechanism and stereochemistry
Prevailing mechanism
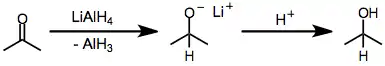
Reduction by alkoxyaluminium hydrides is thought in most cases to proceed by a polar mechanism.[6] Hydride transfer to the organic substrate generates an organic anion, which is neutralized either by protic solvent or upon acidic workup.
-
(2)
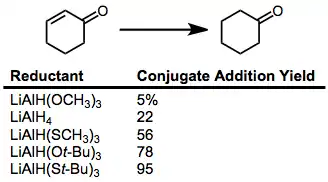
Reductions of α,β-unsaturated carbonyl compounds may occur in a 1,2 sense (direct addition) or a 1,4 sense (conjugate addition). The tendency to add in a 1,4 sense is correlated with the softness of the hydride reagent according to Pearson's hard-soft acid-base theory.[7] Experimental results agree with the theory—softer hydride reagents afford higher yields of the conjugate reduction product.[8]
-
(3)
A few substrates, including diaryl ketones,[9] diarylalkenes,[10] and anthracene,[11] are known to undergo reduction by single-electron transfer pathways with lithium aluminium hydride.
Metal alkoxylaluminium hydride reagents are well characterized in a limited number of cases.[12] Precise characterization is complicated in some cases by disproportionation, which converts alkyoxyaluminium hydrides into alkoxyaluminates and metal aluminium hydride:[13]
-
LiAlHn(OR)4-n ⇌ (4-n) LiAlH4 + n LiAlH(OR)4
(4)
Stereochemistry
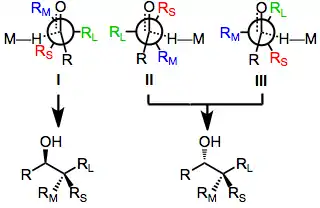
The origin of diastereoselectivity in reductions of chiral ketones has been extensively analyzed and modeled.[14][15] According to a model advanced by Felkin,[16] diastereoselectivity is controlled by the relative energy of the three transition states I, II, and III. Transition state I is favored in the absence of polar groups on the α carbon, and stereoselectivity increases as the size of the achiral ketone substituent (R) increases. Transition state III is favored for reductions of alkyl ketones in which RM is an electron-withdrawing group, because the nucleophile and electron-withdrawing substituent prefer to be as far away from one another as possible.
-
(5)
Diastereoselectivity in reductions of cyclic ketones has also been studied. Conformationally flexible ketones undergo axial attack by the hydride reagent, leading to the equatorial alcohol. Rigid cyclic ketones, on the other hand, undergo primarily equatorial attack to afford the axial alcohol. Preferential equatorial attack on rigid ketones has been rationalized by invoking "steric approach control"—an equatorial approach of the hydride reagent is less sterically hindered than an axial approach.[17] The preference for axial attack on conformationally flexible cyclic ketones has been addressed by a model put forth by Felkin and Anh.[18][19] The transition state for axial attack (IV) suffers from steric strain between any axial substituents and the incoming hydride reagent. The transition state for equatorial attack (V) suffers from torsional strain between the incoming hydride reagent and adjacent equatorial hydrogens. The difference between these two strain energies determines which direction of attack is favored, and when R is small, torsional strain in V dominates and the equatorial alcohol product is favored.
-
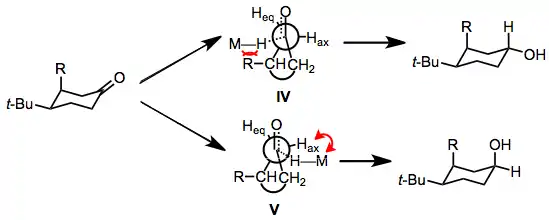
(6)
Scope and limitations
Alkoxyaluminium and closely related hydride reagents reduce a wide variety of functional groups, often with good selectivity. This section, organized by functional group, covers the most common or synthetically useful methods for alkoxyaluminium hydride reduction of organic compounds.
Many selective reductions of carbonyl compounds can be effected by taking advantage of the unique reactivity profiles of metal alkoxylaluminium hydrides. For instance, lithium tri-tert-butoxy)aluminium hydride (LTBA) reduces aldehydes and ketones selectively in the presence of esters, with which it reacts extremely slowly.[20]
-
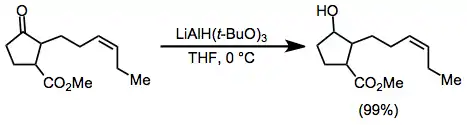
(8)
α,β-Unsaturated ketones may be reduced selectively in a 1,2 or 1,4 sense by a judicious choice of reducing agent. Use of relatively unhindered lithium trimethoxyaluminium hydride results in nearly quantitative direct addition to the carbonyl group (Eq. (9)).[21] On the other hand, use of the bulky reagent LTBA leads to a high yield of the conjugate addition product (Eq. (10)).[22]
-
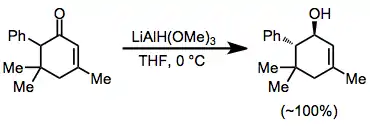
(9)
-
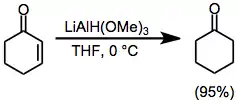
(10)
Ether cleavage is difficult to accomplish with most hydride reagents. However, debenzylation of benzyl aryl ethers may be accomplished with SMEAH.[23] This protocol is a useful alternative to methods requiring acid or hydrogenolysis (e.g., Pd/C and hydrogen gas).
-
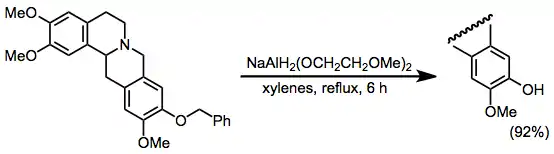
(11)
Epoxides are generally attacked by alkoxyaluminium hydrides at the less substituted position. A nearby hydroxyl group may facilitate intramolecular delivery of the hydride reagent, allowing for selective opening of 1,2-disubstituted epoxides at the position closer to the hydroxyl group.[24] The configuration at the untouched epoxide carbon is preserved.
-
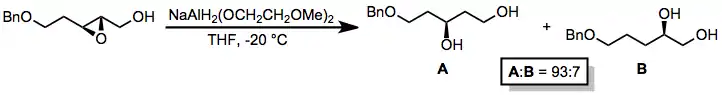
(12)
Unsaturated carbonyl compounds may be reduced either to saturated or unsaturated alcohols by alkoxyaluminium hydride reagents. Addition of an unsaturated aldehyde to a solution of Red-Al afforded the saturated alcohol; inverse addition yielded the unsaturated alcohol product.[25]
-
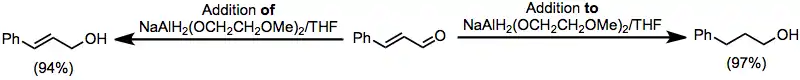
(13)
Alkenes undergo hydroalumination in the presence of some alkoxyaluminium hydrides.[26] In a related application, NaAlH2(OCH2CH2OCH3)2 (sodium bis(methoxyethoxy) aluminium dihydride, SMEAH or Red-Al) reacts with zirconocene dichloride to afford zirconocene chloride hydride (Schwartz's reagent). Alkenes undergo hydrozirconation in the presence of this reagent, affording functionalized products after quenching with an electrophile.[27]
-
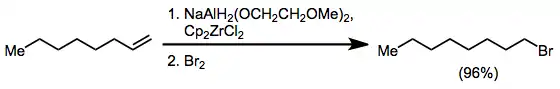
(14)
Functional groups containing heteroatoms other than oxygen may also be reduced to the corresponding hydrocarbons in the presence of an alkoxyaluminium hydride reagent. Primary alkyl halides undergo reduction to the corresponding alkanes in the presence of NaAlH(OH)(OCH2CH2OCH3)2. Secondary halides are less reactive, but afford alkanes in reasonable yield.[28]
-
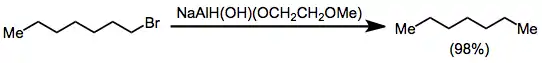
(15)
Sulfoxides are reduced to the corresponding sulfides in good yield in the presence of SMEAH.[29]
-
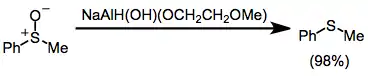
(16)
Imines are reduced by metal alkoxyaluminium hydrides to the corresponding amines. In the example below, use of the exo amine forms with high diastereoselectivity. The selectivity of hydride reduction in this case is higher than that of catalytic hydrogenation.[30]
-
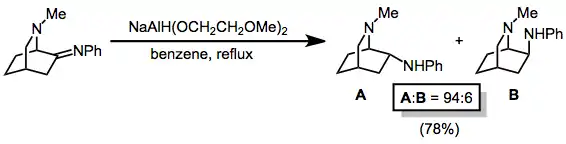
(16)
Experimental conditions and procedure
Preparation of hydride reagents
Alkoxyaluminium hydrides are typically prepared by treatment of lithium aluminium hydride with the corresponding alcohol.[31] Hydrogen evolution indicates the formation of alkoxyaluminium hydride products. Hindered hydrides such as lithium tri-(tert-butoxy)aluminium hydride (LTBA) are stable for long periods of time under inert atmosphere, but lithium trimethoxyaluminium hydride (LTMA) undergoes disproportionation and should be used immediately after preparation. Pure, solid Red-Al is stable for several hours under inert atmosphere and is available commercially as a 70%-solution in toluene under the trade name Vitride or Synhydrid.
Reduction conditions
Reduction may typically be carried out in a round-bottom flask equipped with a drying-tube-capped reflux condenser, a mercury-sealed mechanical stirrer, a thermometer, a nitrogen inlet, and an additional funnel with a pressure-equalizing side arm. The most common solvents used are tetrahydrofuran and diethyl ether. Whatever solvent is used should be anhydrous and pure. Alkoxyaluminium hydrides should be kept as dry as possible and represent a significant fire hazard, particularly when an excess of hydride is used (hydrogen evolves during workup).
Example procedure[32]
-
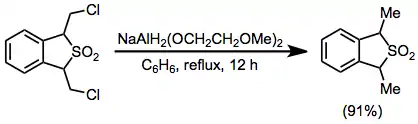
(17)
To a solution of 1,3-dihydro-1,3-bis(chloromethyl)benzo[c] thiophene 2,2-dioxide (0.584 g, 2.2 mmol) in 50 ml of dry benzene was added 0.80 mL (2.8 mmol) of a 70% benzene solution of NaAlH2(OCH2CH2OCH2)2 via syringe, and the solution was refluxed for 12 hours. The mixture was cooled to 0° and decomposed with 20% sulfuric acid. The benzene layer was separated, washed with 10 mL of water, dried over potassium carbonate, and concentrated to give the product as a yellow oil in 91% yield (0.480 g); IR (film) 770, 1140, and 1320 cm–1; NMR (CDCl3) δ 4.22 (q, 2 H), 1.61 and 1.59 (2 d, 6 H, J = 7 Hz), 7.3 (s, 4 H); m/e (rel. intensity) 196 (M+) (14), 132 (M-SO2) (100); MS analysis 196.055796 (calc.), 196.057587 (obs.).
References
- Málek, J. Org. React. 1985, 34, 1. doi:10.1002/0471264180.or034.01
- Málek, J. Org. React. 1988, 36, 249. doi:10.1002/0471264180.or036.03
- Brown, G. Org. React. 1951, 6, 469.
- Schenker, E. in Newer Methods of Preparative Organic Chemistry, Vol. IV., W. Foerst, Ed., Academic Press, New York, 1968, pp. 163–335.
- Itsuno, S. Org. React. 1998, 52, 395.
- House, O. Modern Synthetic Reactions, 2nd ed., W. A. Benjamin, Menlo Park, Calif., 1972.
- Pearson, G. J. Chem. Educ. 1968, 45, 581.
- Bottin, J.; Eisenstein, O.; Minot, C.; Anh, T. Tetrahedron Lett., 1972, 3015.
- Cerný, M.; Málek, J. Collect. Czech. Chem. Commun.. 41, 119 (1976).
- Málek, J.; Cerný, M. J. Organomet. Chem. 1975, 84, 139.
- Málek, J.; Cerný, M.; Rericha, R. Collect. Czech. Chem. Commun. 1974, 39, 2656.
- Bec, M.; Huet, J. Bull. Soc. Chim. Fr., 1972, 1636.
- Brown, C.; Shoaf, J. J. Am. Chem. Soc. 1964, 86, 1079.
- Cram, J.; Abd Elhafez, A. J. Am. Chem. Soc. 1952, 74, 5828.
- Chérest, M.; Prudent, N. Tetrahedron 1980, 36, 1599.
- Chérest, M.; Felkin, H.; Prudent, N. Tetrahedron Lett., 1968, 2199.
- Dauben, W. G.; Fonken, G. J.; Noyce, D. S. J. Am. Chem. Soc. 1956, 78, 2579.
- Chérest, M.; Felkin, H. Tetrahedron Lett., 1971, 383.
- Huet, J.; Maroni-Barnaud, Y.; Anh, N. T.; Seyden-Penne, J. Tetrahedron Lett., 1976, 159.
- Torii, S.; Tanaka, H.; Inokuchi, T.; Tomozane, K. Bull. Chem. Soc. Jpn. 1982, 55, 3947.
- Danh, N. C.; Arnaud, C.; Huet, J. Bull. Soc. Chim. Fr. 1974, 1071.
- Durand, J.; Anh, N. T.; Huet, J. Tetrahedron Lett. 1974, 2397.
- Kametani, T.; Huang, S. P.; Ihara, M.; Fukumoto, K. J. Org. Chem. 1976, 41, 2545.
- Finan, M.; Kishi, Y. Tetrahedron Lett. 1982, 23, 2719.
- Bazant, V.; Capka, M.; Cerny, M.; Chvalovský, V.; Kochloefl, K.; Kraus, M.; Málek, J. Tetrahedron Lett., 1968, 3303.
- Ashby, C.; Noding, A. J. Org. Chem. 1980, 45, 1035.
- Hart, W.; Schwartz, J. J. Am. Chem. Soc. 1974, 96, 8115.
- Capka, M.; Chvalovský, V. Collect. Czech. Chem. Commun. 1969, 34, 3110.
- Weber, L. Chem. Ber. 1983, 116, 2022.
- Law, J.; Lewis, H.; Borne, F. J. Heterocycl. Chem. 1978, 15, 273.
- Véle, I.; Fusek, J.; Štrouf, O. Collect. Czech. Chem. Commun. 1972, 37, 3063.
- Barton, T. J.; Kippenhan, R. C. J. Org. Chem. 1972, 37, 4194.