Alveolar soft part sarcoma
Alveolar soft part sarcoma, abbreviated ASPS, is a very rare type of soft-tissue sarcoma, that grows slowly and whose cell of origin is unknown.
| Alveolar soft part sarcoma | |
|---|---|
| Other names | Alveolar soft-tissue sarcoma |
 | |
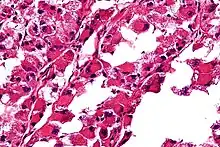
| Micrograph of an alveolar soft part sarcoma, showing the characteristic alveolar-like architecture and cells with eccentric nuclei and abundant eosinophilic cytoplasm. H&E stain. | |
| Specialty | Oncology |
ASPS arises mainly in children and young adults and can migrate (metastasize) into other parts of the body, typically the lungs and the brain. Typically, ASPS arises in muscles and deep soft tissue of the thigh or the leg (lower extremities), but can also appear in the upper extremities (hands, neck, and head). While ASPS is a soft tissue sarcoma, it can also spread and grow inside the bones.
Etymology
- The term alveolar comes from the microscopic pattern, visible during the analysis of slides of ASPS under the microscope in histopathology. The tumor cells seem to be arranged in the same pattern as the cells of the small air sacks (alveoli) in the lungs. However, this is just a structural similarity. ASPS was first described and characterized in 1952.[1]
- ASPS is a sarcoma, and that indicates that this cancer initially arises from tissue of embryonic mesenchymal origin. (The fertilized egg divides and redivides forming a sphere. Early in embryogenesis, dimples appear in the poles of the sphere and burrow through the sphere forming an inner passage that will ultimately form the gut. Malignancies arising from cells that were originally part of the outer layer of the sphere and those that were part of the embryonic tunnel are termed carcinomas; malignancies arising from the cells between the outer layer and the inner burrow are termed sarcomas.)
Causes
Chromosomal analysis of ASPS shows the breaking and joining of two chromosomes in the tumor cells. A piece of chromosome X breaks and is joined to chromosome 17.[2] This translocation creates a fusion between two genes named ASPL and TFE3, which results in the formation of an aberrant protein (termed fusion protein) that is not found in normal cells. Two sorts of fusions between chromosome X and chromosome 17 are found in different ASPS tumors: type one and type two.
Dr. Marc Ladanyi at Memorial Sloan-Kettering Cancer Center, in New York City, has pioneered this work. The resultant fusion protein ASPL–TFE3 is a rogue transcription factor that is the driver of aberrant cellular behavior including uncontrolled cell division and enhanced angiogenesis.
Diagnosis
ASPS may exist in the patient’s body for a long time before being diagnosed. It can grow large and push aside surrounding tissues for a long time before causing any discomfort. Therefore, ASPS symptoms may either be a painless swelling, or a soreness caused by compressed nerves or muscles, affecting the range of motion in the area.
Pathology
The definitive diagnosis of ASPS is based on its appearance under the microscope (i.e., its histomorphology), and presence of the characteristic chromosomal translocation (i.e., cytogenetics).
ASPS' histomorphologic features include an alveolar-like pattern at low magnification and the presence of large cells with abundant eosinophilic cytoplasm and eccentric nuclei. Calcifications are commonly present, as may be seen with slow-growing neoplasms.
Prognosis
Although ASPS displays a relatively indolent course, the ultimate prognosis is poor and is often characterized by late metastases.[3]
Epidemiology
ASPS is an extremely rare cancer. While sarcomas comprise about 1% of all newly diagnosed cancers, and 15% of all childhood cancers, ASPS comprises less than 1% of sarcomas. According to the American Cancer Society, about 9530 new cases of soft tissue sarcoma will be diagnosed in the USA in 2006. This predicts under 100 new cases of ASPS. Such low numbers of occurrence seriously impede the search for a cure by making it hard to gather any meaningful statistics about the disease. As a result, finding the best treatment option often involves making a lot of educated guesses.
Research
- The first xenograft model of ASPS (for type one) was established in mice by David Vistica at the National Cancer Institute in Frederick, MD in 2009.[4] The same authors subsequently generated the first publicly available ASPS cell line (designated ASPS-1).[5]
- An important advance involved demonstrating that the ASPL–TFE3 fusion protein (a transcription factor) enhanced expression of the receptor tyrosine kinase c-MET, making ASPS sensitive to small-molecule kinase inhibitor such as sunitinib.[6][7]
- Current clinical trials are exploring the utility of kinase inhibitors (targeting growth factor pathways and angiogenesis); checkpoint inhibitors and cellular immunotherapies in the treatment of ASPS.[8]
- Researchers at the Huntsman Cancer Institute (HCI) in Utah demonstrated that ASPS might be driven in part by lactate both being used as a fuel and driving angiogenesis.[9]
- In terms of origin, it was recently demonstrated that although ASPS generally arises in muscle tissue, the cells do not express muscle markers at the mRNA level and more closely resemble mesenchymal stromal cells.[10]
References
- Christopherson WM, Foote FW, Stewart FW (1952). "Alveolar soft part sarcomas: structurally characteristic tumors of uncertain histogenesis". Cancer. 1952 (5): 100–111. doi:10.1002/1097-0142(195201)5:1<100::aid-cncr2820050112>3.0.co;2-k. PMID 14886902.
- Ladanyi M, Lui MY, Antonescu CR, Krause-Boehm A, Meindl A, Argani P, et al. (January 2001). "The der(17)t(X;17)(p11;q25) of human alveolar soft part sarcoma fuses the TFE3 transcription factor gene to ASPL, a novel gene at 17q25". Oncogene. 20 (1): 48–57. doi:10.1038/sj.onc.1204074. PMID 11244503.
- Shelke P, Sarode GS, Sarode SC, Anand R, Prajapati G, Patil S (2018). "Alveolar soft-part sarcoma of the oral cavity: A review of literature". Rare Tumors. 10: 2036361318810907. doi:10.1177/2036361318810907. PMC 6299302. PMID 30574289.
- Vistica DT, Hollingshead M, Borgel SD, Kenney S, Stockwin LH, Raffeld M, et al. (August 2009). "Therapeutic vulnerability of an in vivo model of alveolar soft part sarcoma (ASPS) to antiangiogenic therapy". Journal of Pediatric Hematology/Oncology. 31 (8): 561–70. doi:10.1097/MPH.0b013e3181a6e043. PMC 2784654. PMID 19636271.
- Kenney S, Vistica DT, Stockwin LH, Burkett S, Hollingshead MG, Borgel SD, et al. (July 2011). "ASPS-1, a novel cell line manifesting key features of alveolar soft part sarcoma". Journal of Pediatric Hematology/Oncology. 33 (5): 360–8. doi:10.1097/MPH.0b013e3182002f9f. PMC 7518051. PMID 21552147. S2CID 25794748.
- Tsuda M, Davis IJ, Argani P, Shukla N, McGill GG, Nagai M, et al. (February 2007). "TFE3 fusions activate MET signaling by transcriptional up-regulation, defining another class of tumors as candidates for therapeutic MET inhibition". Cancer Research. 67 (3): 919–29. doi:10.1158/0008-5472.CAN-06-2855. PMID 17283122.
- Kummar S, Allen D, Monks A, Polley EC, Hose CD, Ivy SP, et al. (June 2013). "Cediranib for metastatic alveolar soft part sarcoma". Journal of Clinical Oncology. 31 (18): 2296–302. doi:10.1200/JCO.2012.47.4288. PMC 3677840. PMID 23630200.
- Brahmi M, Vanacker H, Dufresne A (July 2020). "Novel therapeutic options for alveolar soft part sarcoma: antiangiogenic therapy, immunotherapy and beyond". Current Opinion in Oncology. 32 (4): 295–300. doi:10.1097/CCO.0000000000000652. PMID 32541316.
- Goodwin ML, Jin H, Straessler K, Smith-Fry K, Zhu JF, Monument MJ, et al. (December 2014). "Modeling alveolar soft part sarcomagenesis in the mouse: a role for lactate in the tumor microenvironment". Cancer Cell. 26 (6): 851–862. doi:10.1016/j.ccell.2014.10.003. PMC 4327935. PMID 25453902.
- Stockwin LH (2020-06-19). "Alveolar soft-part sarcoma (ASPS) resembles a mesenchymal stromal progenitor: evidence from meta-analysis of transcriptomic data". PeerJ. 8: e9394. doi:10.7717/peerj.9394. PMC 7307565. PMID 32596059.