Molecular evolution
Molecular evolution is the process of change in the sequence composition of cellular molecules such as DNA, RNA, and proteins across generations. The field of molecular evolution uses principles of evolutionary biology and population genetics to explain patterns in these changes. Major topics in molecular evolution concern the rates and impacts of single nucleotide changes, neutral evolution vs. natural selection, origins of new genes, the genetic nature of complex traits, the genetic basis of speciation, the evolution of development, and ways that evolutionary forces influence genomic and phenotypic changes.
| Part of a series on |
| Evolutionary biology |
|---|
 |
|
History
The history of molecular evolution starts in the early 20th century with comparative biochemistry, and the use of "fingerprinting" methods such as immune assays, gel electrophoresis, and paper chromatography in the 1950s to explore homologous proteins.[1][2] The field of molecular evolution came into its own in the 1960s and 1970s, following the rise of molecular biology. The advent of protein sequencing allowed molecular biologists to create phylogenies based on sequence comparison, and to use the differences between homologous sequences as a molecular clock to estimate the time since the last universal common ancestor.[1] In the late 1960s, the neutral theory of molecular evolution provided a theoretical basis for the molecular clock,[3] though both the clock and the neutral theory were controversial, since most evolutionary biologists held strongly to panselectionism, with natural selection as the only important cause of evolutionary change. After the 1970s, nucleic acid sequencing allowed molecular evolution to reach beyond proteins to highly conserved ribosomal RNA sequences, the foundation of a reconceptualization of the early history of life.[1]
Forces in molecular evolution
The content and structure of a genome is the product of the molecular and population genetic forces which act upon that genome. Novel genetic variants will arise through mutation and will spread and be maintained in populations due to genetic drift or natural selection.
Genome replication
The ability to undergo self-replication, a fundamental feature of life, is considered to have emerged when a molecule similar to a double-stranded polynucleotide (possibly similar to RNA) dissociated into single-stranded polynucleotides and each of these served as a template for synthesis of a complementary strand producing two double stranded copies.[4] In such a system, individual replicators with different nucleotide sequences could compete with each other for nucleotide resources, thus initiating natural selection for the most “fit” sequences.[4] In this initially inaccurate replication system, mutations would naturally arise that influence the folding state of the polynucleotides and thus affect the propensities for strand association (promoting stability) and disassociation (allowing genome replication).
Mutation

Mutations are permanent, transmissible changes to the genetic material (DNA or RNA) of a cell or virus. Mutations result from errors in DNA replication during cell division and by exposure to radiation, chemicals, and other environmental stressors, or viruses and transposable elements. Point mutations such as single-nucleotide substitutions, which modify just one base-pair of the DNA, are often the most common class of mutations. Other types of mutations modify larger segments of DNA and can cause duplications, insertions, deletions, inversions, and translocations.
The distribution of rates for diverse kinds of mutations (across the entire genome, or in some region of interest) is called the "mutation spectrum" (for a general treatment, see App. B of [5]). Mutations of different types occur at widely varying rates. Perhaps the most common type of mutation in humans is a change in the length of a Variable_number_tandem_repeat (e.g., the CAG repeats underlying various disease-associated mutations). Such STR mutations may occur at rates on the order of 10-3 per generation,[6] whereas most types of mutations occur at much lower rates. Even within a class of mutations, there are strong biases, e.g., Transitions (A ↔ G or C ↔ T) are more common than transversions (purine (adenine or guanine)) ↔ pyrimidine (cytosine or thymine, or in RNA, uracil))[7]. These mutational tendencies can play an important role in evolution via bias in the introduction of variation (arrival bias), contributing to parallelism, trends, and differences in the navigability of adaptive landscapes.[8]
Mutations are stochastic and typically occur randomly across genes. Mutation rates for single nucleotide sites for most organisms are very low, roughly 10−9 to 10−8 per site per generation, though some viruses have higher mutation rates on the order of 10−6 per site per generation. Among these mutations, some will be neutral or beneficial and will remain in the genome unless lost via genetic drift, and others will be detrimental and will be eliminated from the genome by natural selection.
Because mutations are extremely rare, they accumulate very slowly across generations. While the number of mutations which appears in any single generation may vary, over very long time periods they will appear to accumulate at a regular pace. Using the mutation rate per generation and the number of nucleotide differences between two sequences, divergence times can be estimated effectively via the molecular clock.
Recombination

Recombination is a process that results in genetic exchange between chromosomes or chromosomal regions (see Figure). Recombination counteracts physical linkage between adjacent genes, thereby reducing genetic hitchhiking. The resulting independent inheritance of genes results in more efficient selection, meaning that regions with higher recombination will harbor fewer detrimental mutations, more selectively favored variants, and fewer errors in replication and repair. Recombination can also generate particular types of mutations if chromosomes are misaligned.
Gene conversion
Gene conversion is a type of recombination that is the product of DNA repair where nucleotide damage is corrected using an homologous genomic region as a template. Damaged bases are first excised, the damaged strand is then aligned with an undamaged homologous strand, and DNA synthesis repairs the excised region using the undamaged strand as a guide. Gene conversion is often responsible for homogenizing sequences of duplicate genes over long time periods, reducing nucleotide divergence.
Genetic drift
Genetic drift is the change of allele frequencies from one generation to the next due to stochastic effects of random sampling in finite populations. Some existing variants have no effect on fitness and may increase or decrease in frequency simply due to chance. "Nearly neutral" variants whose selection coefficient is close to a threshold value of 1 / the effective population size will also be affected by chance as well as by selection and mutation. Many genomic features have been ascribed to accumulation of nearly neutral detrimental mutations as a result of small effective population sizes.[9] With a smaller effective population size, a larger variety of mutations will behave as if they are neutral due to inefficiency of selection.
Selection
Selection occurs when organisms with greater fitness, i.e. greater ability to survive or reproduce, are favored in subsequent generations, thereby increasing the instance of underlying genetic variants in a population. Selection can be the product of natural selection, artificial selection, or sexual selection. Natural selection is any selective process that occurs due to the fitness of an organism to its environment. In contrast sexual selection is a product of mate choice and can favor the spread of genetic variants which act counter to natural selection but increase desirability to the opposite sex or increase mating success. Artificial selection, also known as selective breeding, is imposed by an outside entity, typically humans, in order to increase the frequency of desired traits.
The principles of population genetics apply similarly to all types of selection, though in fact each may produce distinct effects due to clustering of genes with different functions in different parts of the genome, or due to different properties of genes in particular functional classes. For instance, sexual selection could be more likely to affect molecular evolution of the sex chromosomes due to clustering of sex specific genes on the X, Y, Z or W.
Intragenomic conflict
Selection can operate at the gene level at the expense of organismal fitness, resulting in intragenomic conflict. This is because there can be a selective advantage for selfish genetic elements in spite of a host cost. Examples of such selfish elements include transposable elements, meiotic drivers, killer X chromosomes, selfish mitochondria, and self-propagating introns.
Genome architecture
Genome size
Genome size is influenced by the amount of repetitive DNA as well as number of genes in an organism. The C-value paradox refers to the lack of correlation between organism 'complexity' and genome size. Explanations for the so-called paradox are two-fold. First, repetitive genetic elements can comprise large portions of the genome for many organisms, thereby inflating DNA content of the haploid genome. Secondly, the number of genes is not necessarily indicative of the number of developmental stages or tissue types in an organism. An organism with few developmental stages or tissue types may have large numbers of genes that influence non-developmental phenotypes, inflating gene content relative to developmental gene families.
Neutral explanations for genome size suggest that when population sizes are small, many mutations become nearly neutral. Hence, in small populations repetitive content and other 'junk' DNA can accumulate without placing the organism at a competitive disadvantage. There is little evidence to suggest that genome size is under strong widespread selection in multicellular eukaryotes. Genome size, independent of gene content, correlates poorly with most physiological traits and many eukaryotes, including mammals, harbor very large amounts of repetitive DNA.
However, birds likely have experienced strong selection for reduced genome size, in response to changing energetic needs for flight. Birds, unlike humans, produce nucleated red blood cells, and larger nuclei lead to lower levels of oxygen transport. Bird metabolism is far higher than that of mammals, due largely to flight, and oxygen needs are high. Hence, most birds have small, compact genomes with few repetitive elements. Indirect evidence suggests that non-avian theropod dinosaur ancestors of modern birds[10] also had reduced genome sizes, consistent with endothermy and high energetic needs for running speed. Many bacteria have also experienced selection for small genome size, as time of replication and energy consumption are so tightly correlated with fitness.
Repetitive elements
Transposable elements are self-replicating, selfish genetic elements which are capable of proliferating within host genomes. Many transposable elements are related to viruses, and share several proteins in common....
Chromosome number and organization
The number of chromosomes in an organism's genome also does not necessarily correlate with the amount of DNA in its genome. The ant Myrmecia pilosula has only a single pair of chromosomes[11] whereas the Adders-tongue fern Ophioglossum reticulatum has up to 1260 chromosomes.[12] Cilliate genomes house each gene in individual chromosomes, resulting in a genome which is not physically linked. Reduced linkage through creation of additional chromosomes should effectively increase the efficiency of selection.
Changes in chromosome number can play a key role in speciation, as differing chromosome numbers can serve as a barrier to reproduction in hybrids. Human chromosome 2 was created from a fusion of two chimpanzee chromosomes and still contains central telomeres as well as a vestigial second centromere. Polyploidy, especially allopolyploidy, which occurs often in plants, can also result in reproductive incompatibilities with parental species. Agrodiatus blue butterflies have diverse chromosome numbers ranging from n=10 to n=134 and additionally have one of the highest rates of speciation identified to date.[13]
Gene content and distribution
Different organisms house different numbers of genes within their genomes as well as different patterns in the distribution of genes throughout the genome. Some organisms, such as most bacteria, Drosophila, and Arabidopsis have particularly compact genomes with little repetitive content or non-coding DNA. Other organisms, like mammals or maize, have large amounts of repetitive DNA, long introns, and substantial spacing between different genes. The content and distribution of genes within the genome can influence the rate at which certain types of mutations occur and can influence the subsequent evolution of different species. Genes with longer introns are more likely to recombine due to increased physical distance over the coding sequence. As such, long introns may facilitate ectopic recombination, and result in higher rates of new gene formation.
Organelles
In addition to the nuclear genome, endosymbiont organelles contain their own genetic material typically as circular plasmids. Mitochondrial and chloroplast DNA varies across taxa, but membrane-bound proteins, especially electron transport chain constituents are most often encoded in the organelle. Chloroplasts and mitochondria are maternally inherited in most species, as the organelles must pass through the egg. In a rare departure, some species of mussels are known to inherit mitochondria from father to son.
Origins of new genes
New genes arise from several different genetic mechanisms including gene duplication, de novo origination, retrotransposition, chimeric gene formation, recruitment of non-coding sequence, and gene truncation.
Gene duplication initially leads to redundancy. However, duplicated gene sequences can mutate to develop new functions or specialize so that the new gene performs a subset of the original ancestral functions. In addition to duplicating whole genes, sometimes only a domain or part of a protein is duplicated so that the resulting gene is an elongated version of the parental gene.
Retrotransposition creates new genes by copying mRNA to DNA and inserting it into the genome. Retrogenes often insert into new genomic locations, and often develop new expression patterns and functions.
Chimeric genes form when duplication, deletion, or incomplete retrotransposition combine portions of two different coding sequences to produce a novel gene sequence. Chimeras often cause regulatory changes and can shuffle protein domains to produce novel adaptive functions.
De novo gene birth can also give rise to new genes from previously non-coding DNA.[14] For instance, Levine and colleagues reported the origin of five new genes in the D. melanogaster genome from noncoding DNA.[15][16] Similar de novo origin of genes has been also shown in other organisms such as yeast,[17] rice[18] and humans.[19] De novo genes may evolve from transcripts that are already expressed at low levels.[20] Mutation of a stop codon to a regular codon or a frameshift may cause an extended protein that includes a previously non-coding sequence. The formation of novel genes from scratch typically can not occur within genomic regions of high gene density. The essential events for de novo formation of genes is recombination/mutation which includes insertions, deletions, and inversions. These events are tolerated if the consequence of these genetic events does not interfere in cellular activities. Most genomes comprise prophages wherein genetic modifications do not, in general, affect the host genome propagation. Hence, there is higher probability of genetic modifications, in regions such as prophages, which is proportional to the probability of de novo formation of genes.[21]
De novo evolution of genes can also be simulated in the laboratory. For example, semi-random gene sequences can be selected for specific functions.[22] More specifically, they selected sequences from a library that could complement a gene deletion in E. coli. The deleted gene encodes ferric enterobactin esterase (Fes), which releases iron from an iron chelator, enterobactin. While Fes is a 400 amino acid protein, the newly selected gene was only 100 amino acids in length and unrelated in sequence to Fes.[22] A similar approach has been used to select for random peptides and short proteins that can compensate for the lack of an essential enzyme, SerB, in E. coli. Indeed, such random proteins with a selective benefit can be created and thus provide evidence for evolution of functional proteins from non-functional sequences.[23]
In vitro molecular evolution experiments
Principles of molecular evolution can be discovered and tested using laboratory experimentation. This usually involves the cloning and in vitro modification of genes and proteins outside cells. Since the pioneering work of Sol Spiegelmann in 1967 [ref], involving RNA that replicates itself with the aid of an enzyme extracted from the Qß virus [ref], several groups (such as Kramers [ref] and Biebricher/Luce/Eigen [ref]) studied mini and micro variants of this RNA in the 1970s and 1980s that replicate on the timescale of seconds to a minute, allowing hundreds of generations with large population sizes (e.g. 10^14 sequences) to be followed in a single day of experimentation. The chemical kinetic elucidation of the detailed mechanism of replication [ref, ref] meant that this type of system was the first molecular evolution system that could be fully characterised on the basis of physical chemical kinetics, later allowing the first models of the genotype to phenotype map based on sequence dependent RNA folding and refolding to be produced [ref, ref]. Subject to maintaining the function of the multicomponent Qß enzyme, chemical conditions could be varied significantly, in order to study the influence of changing environments and selection pressures [ref]. Experiments with in vitro RNA quasi species included the characterisation of the error threshold for information in molecular evolution [ref], the discovery of de novo evolution [ref] leading to diverse replicating RNA species and the discovery of spatial travelling waves as ideal molecular evolution reactors [ref, ref]. Later experiments employed novel combinations of enzymes to elucidate novel aspects of interacting molecular evolution involving population dependent fitness, including work with artificially designed molecular predator prey and cooperative systems of multiple RNA and DNA [ref, ref]. Special evolution reactors were designed for these studies, starting with serial transfer machines, flow reactors such as cell-stat machines, capillary reactors, and microreactors including line flow reactors and gel slice reactors. These studies were accompanied by theoretical developments and simulations involving RNA folding and replication kinetics that elucidated the importance of the correlation structure between distance in sequence space and fitness changes [ref], including the role of neutral networks and structural ensembles in evolutionary optimisation.
in vitro protein function evolution
Mutagenic hot spots in enzymes can be identified using NMR spectroscopy. In a proof-of-concept study, Bhattacharya and colleagues converted myoglobin, a non-enzymatic oxygen storage protein, into a highly efficient Kemp eliminase using only three mutations. This demonstrates that only few mutations are needed to radically change the function of a protein.[24]
Molecular phylogenetics
Molecular systematics is the product of the traditional fields of systematics and molecular genetics.[25] It uses DNA, RNA, or protein sequences to resolve questions in systematics, i.e. about their correct scientific classification or taxonomy from the point of view of evolutionary biology.
Molecular systematics has been made possible by the availability of techniques for DNA sequencing, which allow the determination of the exact sequence of nucleotides or bases in either DNA or RNA. At present it is still a long and expensive process to sequence the entire genome of an organism, and this has been done for only a few species. However, it is quite feasible to determine the sequence of a defined area of a particular chromosome. Typical molecular systematic analyses require the sequencing of around 1000 base pairs.
The driving forces of evolution
Depending on the relative importance assigned to the various forces of evolution, three perspectives provide evolutionary explanations for molecular evolution.[26][27]
Selectionist hypotheses argue that selection is the driving force of molecular evolution. While acknowledging that many mutations are neutral, selectionists attribute changes in the frequencies of neutral alleles to linkage disequilibrium with other loci that are under selection, rather than to random genetic drift.[28] Biases in codon usage are usually explained with reference to the ability of even weak selection to shape molecular evolution.[29]
Neutralist hypotheses emphasize the importance of mutation, purifying selection, and random genetic drift.[30] The introduction of the neutral theory by Kimura,[31] quickly followed by King and Jukes' own findings,[32] led to a fierce debate about the relevance of neodarwinism at the molecular level. The Neutral theory of molecular evolution proposes that most mutations in DNA are at locations not important to function or fitness. These neutral changes drift towards fixation within a population. Positive changes will be very rare, and so will not greatly contribute to DNA polymorphisms.[33] Deleterious mutations do not contribute much to DNA diversity because they negatively affect fitness and so are removed from the gene pool before long.[34] This theory provides a framework for the molecular clock.[33] The fate of neutral mutations are governed by genetic drift, and contribute to both nucleotide polymorphism and fixed differences between species.[35][36]
Some researchers argue that the neutral theory is not accurate and they mostly focus on the rates of positive effects of even small evolutionary changes.[37] They point out that subtle changes in DNA very often have effects, but sometimes these effects are too small for natural selection to act on.[37] Synonymous mutations are not necessarily neutral[37] because there is not a uniform amount of each codon. The nearly neutral theory expanded the neutralist perspective, suggesting that several mutations are nearly neutral, which means both random drift and natural selection is relevant to their dynamics.[37] The main difference between the neutral theory and nearly neutral theory is that the latter focuses on weak selection, not strictly neutral.[34]
Another concept is constructive neutral evolution (CNE), which explains that complex systems can emerge and spread into a population through neutral transitions with the principles of excess capacity, presuppression, and ratcheting,[38][39][40] and it has been applied in areas ranging from the origins of the spliceosome to the complex interdependence of microbial communities.[41][42][43]
Mutationist hypotheses emphasize the dependence of evolution on distinctive mutations,[44] and the systematic or predictable contributions of biases in mutation to parallel evolution or trends.[5] Freese [45] and Sueoka [46] were the first to suggest that genomic GC content and proteome composition might reflect mutational tendencies. Mutational explanations for broad patterns such as codon usage bias are often considered in molecular evolution (see [47]). Although such hypotheses are often associated with neutrality, recent theoretical and empirical results have established that mutational tendencies can influence both neutral and adaptive evolution via bias in the introduction of variation (arrival bias).
Protein evolution
While genomes store information and accumulate mutations, proteins are the active products of genes. Hence the evolution of protein function is critical to understand molecular evolution.
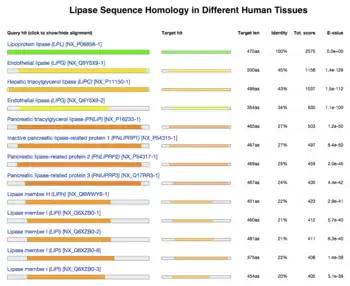
Evolution of proteins is studied by comparing the sequences and structures of proteins from many organisms. Similar sequences/structures indicating that the proteins diverged from a common origin; these proteins are homologous. Phylogenetic analysis of proteins has revealed how proteins evolve and change their structure and function over time.[48][49]
Evolutionary rate. Using the amino acid sequences of hemoglobin and cytochrome c from multiple species, scientists were able to derive estimations of protein evolution rates.[34] Each protein has its own rate, and that rate is relatively constant across phylogenies (e.g., hemoglobin does not evolve at the same rate as cytochrome c, but hemoglobins from humans, mice, etc. do have comparable rates of evolution). Not all regions within a protein mutate at the same rate; functionally important areas mutate more slowly and amino acid substitutions involving similar amino acids occurs more often than dissimilar substitutions.[34] Overall, the level of polymorphisms in proteins seems to be fairly constant. Several species (including humans, fruit flies, and mice) have similar levels of protein polymorphism.[33]
Functional evolution. Numerous enzymes and other proteins have been shown to change their function over the course of evolution. For example, ribonucleotide reductase (RNR) is known from thousands of organisms and has evolved a multitude of structural and functional variants. Class I RNRs use a ferritin subunit and differ by the metal they use as cofactors. In class II RNRs, the thiyl radical is generated using an adenosylcobalamin cofactor and these enzymes do not require additional subunits (as opposed to class I which do). In class III RNRs, the thiyl radical is generated using S-adenosylmethionine bound to a [4Fe-4S] cluster. That is, within a single family of proteins numerous structural and functional mechanisms can evolve.[50]
Functional constructivism principle was used to explain the evolution of neurotransmitters' and hormonal differential regulation of behaviour.[51][52]
Relation to nucleic acid evolution
Protein evolution is inescapably tied to changes and selection of DNA polymorphisms and mutations because protein sequences change in response to alterations in the DNA sequence. Amino acid sequences and nucleic acid sequences do not mutate at the same rate. Due to the degenerate nature of DNA, bases can change without affecting the amino acid sequence. For example, there are six codons that code for leucine. Thus, despite the difference in mutation rates, it is essential to incorporate nucleic acid evolution into the discussion of protein evolution. At the end of the 1960s, two groups of scientists—Kimura (1968) and King and Jukes (1969)—independently proposed that a majority of the evolutionary changes observed in proteins were neutral.[33][34] Since then, the neutral theory has been expanded upon and debated.[34]
Discordance with morphological evolution
There are sometimes discordances between molecular and morphological evolution, which are reflected in molecular and morphological systematic studies, especially of bacteria, archaea and eukaryotic microbes. These discordances can be categorized as two types: (i) one morphology, multiple lineages (e.g. morphological convergence, cryptic species) and (ii) one lineage, multiple morphologies (e.g. phenotypic plasticity, multiple life-cycle stages). Neutral evolution possibly could explain the incongruences in some cases.[53]
Journals and societies
The Society for Molecular Biology and Evolution publishes the journals "Molecular Biology and Evolution" and "Genome Biology and Evolution" and holds an annual international meeting. Other journals dedicated to molecular evolution include Journal of Molecular Evolution and Molecular Phylogenetics and Evolution. Research in molecular evolution is also published in journals of genetics, molecular biology, genomics, systematics, and evolutionary biology.
See also
- Abiogenesis
- Adaptor protein evolution
- Comparative phylogenetics
- Evolution
- E. coli long-term evolution experiment
- Evolutionary physiology
- Evolution of dietary antioxidants
- Genomic organization
- Genetic drift
- Genome evolution
- Heterotachy
- History of molecular evolution
- Horizontal gene transfer
- Human evolution
- Molecular clock
- Molecular paleontology
- Neutral theory of molecular evolution
- Nucleotide diversity
- Parsimony
- Population genetics
- Selection
References
- Dietrich MR (1998). "Paradox and persuasion: negotiating the place of molecular evolution within evolutionary biology". Journal of the History of Biology. 31 (1): 85–111. doi:10.1023/A:1004257523100. PMID 11619919. S2CID 29935487.
- Hagen JB (1999). "Naturalists, molecular biologists, and the challenges of molecular evolution". Journal of the History of Biology. 32 (2): 321–341. doi:10.1023/A:1004660202226. PMID 11624208. S2CID 26994015.
- King JL, Jukes TH (May 1969). "Non-Darwinian evolution". Science. 164 (3881): 788–798. Bibcode:1969Sci...164..788L. doi:10.1126/science.164.3881.788. PMID 5767777.
- HenryQuastler (1964) Emergence of Biological Organization, Yale University Press, New Haven Connecticut ASIN: B0000CMHJ2
- A. Stoltzfus (2021). Mutation, Randomness and Evolution. Oxford, Oxford.
- J. L. Weber and C. Wong (1993). "Mutation of human short tandem repeats". Hum Mol Genet. 2 (8): 1123–8. doi:10.1093/hmg/2.8.1123. PMID 8401493.
- "Transitions vs transversions".
- A. V. Cano and J. L. Payne (2020). "Mutation bias interacts with composition bias to influence adaptive evolution". PLOS Computational Biology. 16 (9): e1008296. Bibcode:2020PLSCB..16E8296C. doi:10.1371/journal.pcbi.1008296. PMC 7571706. PMID 32986712.
- Lynch M (2007). The Origins of Genome Architecture. Sinauer. ISBN 978-0-87893-484-3.
- Organ CL, Shedlock AM, Meade A, Pagel M, Edwards SV (March 2007). "Origin of avian genome size and structure in non-avian dinosaurs". Nature. 446 (7132): 180–184. Bibcode:2007Natur.446..180O. doi:10.1038/nature05621. PMID 17344851. S2CID 3031794.
- Crosland MW, Crozier RH (March 1986). "Myrmecia pilosula, an Ant with Only One Pair of Chromosomes". Science. 231 (4743): 1278. Bibcode:1986Sci...231.1278C. doi:10.1126/science.231.4743.1278. PMID 17839565. S2CID 25465053.
- Gerardus J. H. Grubben (2004). Vegetables. PROTA. p. 404. ISBN 978-90-5782-147-9. Retrieved 10 March 2013.
- Kandul NP, Lukhtanov VA, Pierce NE (March 2007). "Karyotypic diversity and speciation in Agrodiaetus butterflies". Evolution; International Journal of Organic Evolution. 61 (3): 546–559. doi:10.1111/j.1558-5646.2007.00046.x. PMID 17348919.
- McLysaght A, Guerzoni D (September 2015). "New genes from non-coding sequence: the role of de novo protein-coding genes in eukaryotic evolutionary innovation". Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences. 370 (1678): 20140332. doi:10.1098/rstb.2014.0332. PMC 4571571. PMID 26323763.
- Levine MT, Jones CD, Kern AD, Lindfors HA, Begun DJ (June 2006). "Novel genes derived from noncoding DNA in Drosophila melanogaster are frequently X-linked and exhibit testis-biased expression". Proceedings of the National Academy of Sciences of the United States of America. 103 (26): 9935–9939. Bibcode:2006PNAS..103.9935L. doi:10.1073/pnas.0509809103. PMC 1502557. PMID 16777968.
- Zhou Q, Zhang G, Zhang Y, Xu S, Zhao R, Zhan Z, et al. (September 2008). "On the origin of new genes in Drosophila". Genome Research. 18 (9): 1446–1455. doi:10.1101/gr.076588.108. PMC 2527705. PMID 18550802.
- Cai J, Zhao R, Jiang H, Wang W (May 2008). "De novo origination of a new protein-coding gene in Saccharomyces cerevisiae". Genetics. 179 (1): 487–496. doi:10.1534/genetics.107.084491. PMC 2390625. PMID 18493065.
- Xiao W, Liu H, Li Y, Li X, Xu C, Long M, Wang S (2009). El-Shemy HA (ed.). "A rice gene of de novo origin negatively regulates pathogen-induced defense response". PLOS ONE. 4 (2): e4603. Bibcode:2009PLoSO...4.4603X. doi:10.1371/journal.pone.0004603. PMC 2643483. PMID 19240804.
- Knowles DG, McLysaght A (October 2009). "Recent de novo origin of human protein-coding genes". Genome Research. 19 (10): 1752–1759. doi:10.1101/gr.095026.109. PMC 2765279. PMID 19726446.
- Wilson BA, Masel J (2011). "Putatively noncoding transcripts show extensive association with ribosomes". Genome Biology and Evolution. 3: 1245–1252. doi:10.1093/gbe/evr099. PMC 3209793. PMID 21948395.
- Ramisetty BC, Sudhakari PA (2019). "Bacterial 'Grounded' Prophages: Hotspots for Genetic Renovation and Innovation". Frontiers in Genetics. 10: 65. doi:10.3389/fgene.2019.00065. PMC 6379469. PMID 30809245.
- Donnelly AE, Murphy GS, Digianantonio KM, Hecht MH (March 2018). "A de novo enzyme catalyzes a life-sustaining reaction in Escherichia coli". Nature Chemical Biology. 14 (3): 253–255. doi:10.1038/nchembio.2550. PMID 29334382.
- Babina, Arianne M; Surkov, Serhiy; Ye, Weihua; Jerlström-Hultqvist, Jon; Larsson, Mårten; Holmqvist, Erik; Jemth, Per; Andersson, Dan I; Knopp, Michael (2023-03-15). Wade, Joseph T (ed.). "Rescue of Escherichia coli auxotrophy by de novo small proteins". eLife. 12: e78299. doi:10.7554/eLife.78299. ISSN 2050-084X. PMC 10065794. PMID 36920032.
- Bhattacharya S, Margheritis EG, Takahashi K, Kulesha A, D'Souza A, Kim I, et al. (October 2022). "NMR-guided directed evolution". Nature. 610 (7931): 389–393. Bibcode:2022Natur.610..389B. doi:10.1038/s41586-022-05278-9. PMC 10116341. PMID 36198791. S2CID 245067145.
- Lewis-Oritt N, Porter CA, Baker RJ (September 2001). "Molecular systematics of the family Mormoopidae (Chiroptera) based on cytochrome b and recombination activating gene 2 sequences". Molecular Phylogenetics and Evolution. 20 (3): 426–436. doi:10.1006/mpev.2001.0978. PMID 11527468.
- Graur D, Li WH (2000). Fundamentals of molecular evolution. Sinauer. ISBN 0-87893-266-6.
- Casillas S, Barbadilla A (March 2017). "Molecular Population Genetics". Genetics. 205 (3): 1003–1035. doi:10.1534/genetics.116.196493. PMC 5340319. PMID 28270526.
- Hahn MW (February 2008). "Toward a selection theory of molecular evolution". Evolution; International Journal of Organic Evolution. 62 (2): 255–265. doi:10.1111/j.1558-5646.2007.00308.x. PMID 18302709.
- Hershberg R, Petrov DA (December 2008). "Selection on codon bias". Annual Review of Genetics. 42 (1): 287–299. doi:10.1146/annurev.genet.42.110807.091442. PMID 18983258. S2CID 7085012.
- Kimura, M. (1983). The Neutral Theory of Molecular Evolution. Cambridge University Press, Cambridge. ISBN 0-521-23109-4.
- Kimura M (February 1968). "Evolutionary rate at the molecular level". Nature. 217 (5129): 624–626. Bibcode:1968Natur.217..624K. doi:10.1038/217624a0. PMID 5637732. S2CID 4161261.
- King JL, Jukes TH (May 1969). "Non-Darwinian evolution". Science. 164 (3881): 788–798. Bibcode:1969Sci...164..788L. doi:10.1126/science.164.3881.788. PMID 5767777.
- Akashi H, Osada N, Ohta T (September 2012). "Weak selection and protein evolution". Genetics. 192 (1): 15–31. doi:10.1534/genetics.112.140178. PMC 3430532. PMID 22964835.
- Fay JC, Wu CI (2003). "Sequence divergence, functional constraint, and selection in protein evolution". Annual Review of Genomics and Human Genetics. 4: 213–235. doi:10.1146/annurev.genom.4.020303.162528. PMID 14527302. S2CID 6360375.
- Nachman M (2006). "Detecting selection at the molecular level". In Fox CW, Wolf JB (eds.). Evolutionary Genetics: concepts and case studies. pp. 103–118.
- The nearly neutral theory expanded the neutralist perspective, suggesting that several mutations are nearly neutral, which means both random drift and natural selection is relevant to their dynamics.
- Ohta T (1992). "The Nearly Neutral Theory of Molecular Evolution". Annual Review of Ecology and Systematics. 23 (1): 263–286. doi:10.1146/annurev.es.23.110192.001403. ISSN 0066-4162.
- Stoltzfus A (August 1999). "On the possibility of constructive neutral evolution". Journal of Molecular Evolution. 49 (2): 169–181. Bibcode:1999JMolE..49..169S. doi:10.1007/PL00006540. PMID 10441669. S2CID 1743092.
- Stoltzfus A (October 2012). "Constructive neutral evolution: exploring evolutionary theory's curious disconnect". Biology Direct. 7 (1): 35. doi:10.1186/1745-6150-7-35. PMC 3534586. PMID 23062217.
- Muñoz-Gómez SA, Bilolikar G, Wideman JG, Geiler-Samerotte K (April 2021). "Constructive Neutral Evolution 20 Years Later". Journal of Molecular Evolution. 89 (3): 172–182. Bibcode:2021JMolE..89..172M. doi:10.1007/s00239-021-09996-y. PMC 7982386. PMID 33604782.
- Lukeš J, Archibald JM, Keeling PJ, Doolittle WF, Gray MW (July 2011). "How a neutral evolutionary ratchet can build cellular complexity". IUBMB Life. 63 (7): 528–537. doi:10.1002/iub.489. PMID 21698757. S2CID 7306575.
- Vosseberg J, Snel B (December 2017). "Domestication of self-splicing introns during eukaryogenesis: the rise of the complex spliceosomal machinery". Biology Direct. 12 (1): 30. doi:10.1186/s13062-017-0201-6. PMC 5709842. PMID 29191215.
- Brunet TD, Doolittle WF (19 March 2018). "The generality of Constructive Neutral Evolution". Biology & Philosophy. 33 (1): 2. doi:10.1007/s10539-018-9614-6. ISSN 1572-8404. S2CID 90290787.
- M. Nei (2013). Mutation-Driven Evolution. Oxford University Press.
- E. Freese (1962). "On the Evolution of the Base Composition of DNA". J. Theoret. Biol. 3 (1): 82–101. Bibcode:1962JThBi...3...82F. doi:10.1016/S0022-5193(62)80005-8.
It is unimportant in this connection whether selection has been negligible or self-cancelling.
- N. Sueoka (1962). "On the Genetic Basis of Variation and Heterogeneity of DNA Base Composition". Proc. Natl. Acad. Sci. U.S.A. 48 (4): 582–592. Bibcode:1962PNAS...48..582S. doi:10.1073/pnas.48.4.582. PMC 220819. PMID 13918161.
- A. Stoltzfus and L. Y. Yampolsky (2009). "Climbing mount probable: mutation as a cause of nonrandomness in evolution". J Hered. 100 (5): 637–47. doi:10.1093/jhered/esp048. PMID 19625453.
- Hanukoglu I (February 2017). "ASIC and ENaC type sodium channels: conformational states and the structures of the ion selectivity filters". The FEBS Journal. 284 (4): 525–545. doi:10.1111/febs.13840. PMID 27580245. S2CID 24402104.
- Hanukoglu I, Hanukoglu A (April 2016). "Epithelial sodium channel (ENaC) family: Phylogeny, structure-function, tissue distribution, and associated inherited diseases". Gene. 579 (2): 95–132. doi:10.1016/j.gene.2015.12.061. PMC 4756657. PMID 26772908.
- Burnim AA, Spence MA, Xu D, Jackson CJ, Ando N (September 2022). Ben-Tal N, Weigel D, Ben-Tal N, Stubbe J, Hofer A (eds.). "Comprehensive phylogenetic analysis of the ribonucleotide reductase family reveals an ancestral clade". eLife. 11: e79790. doi:10.7554/eLife.79790. PMC 9531940. PMID 36047668.
- Trofimova, I. (2021). "Contingent tunes of neurochemical ensembles in the norm and pathology: can we see the patterns?". Neuropsychobiology. 80 (2): 101–133. doi:10.1159/000513688. PMID 33721867. S2CID 232243254.
- Trofimova, I (2021). "Functional constructivism approach to multilevel nature of biobehavioural diversity". Frontiers in Psychiatry. 12: 641286. doi:10.3389/fpsyt.2021.641286. PMC 8578849. PMID 34777031.
- Lahr DJ, Laughinghouse HD, Oliverio AM, Gao F, Katz LA (October 2014). "How discordant morphological and molecular evolution among microorganisms can revise our notions of biodiversity on Earth". BioEssays. 36 (10): 950–959. doi:10.1002/bies.201400056. PMC 4288574. PMID 25156897.
Further reading
- Li WH (2006). Molecular Evolution. Sinauer. ISBN 0-87893-480-4.
- Lynch M (2007). The Origins of Genome Architecture. Sinauer. ISBN 978-0-87893-484-3.
- Meyer A, van de Peer Y, eds. (2003). Genome evolution : gene and genome duplications and the origin of novel gene functions. Dordrecht: Kluwer Academic Pub. ISBN 978-1-4020-1021-7.
- Gregory TR (2005). The evolution of the genome. Burlington, MA: Elsevier Academic. ISBN 978-0-12-301463-4.
- Levinson G (2020). Rethinking evolution : the revolution that's hiding in plain sight. London: World Scientific. ISBN 978-1-78634-726-8.