Single particle analysis
Single particle analysis is a group of related computerized image processing techniques used to analyze images from transmission electron microscopy (TEM).[1] These methods were developed to improve and extend the information obtainable from TEM images of particulate samples, typically proteins or other large biological entities such as viruses. Individual images of stained or unstained particles are very noisy, and so hard to interpret. Combining several digitized images of similar particles together gives an image with stronger and more easily interpretable features. An extension of this technique uses single particle methods to build up a three-dimensional reconstruction of the particle. Using cryo-electron microscopy it has become possible to generate reconstructions with sub-nanometer resolution and near-atomic resolution[2][3] first in the case of highly symmetric viruses, and now in smaller, asymmetric proteins as well.[4] Single particle analysis can also be performed by induced coupled plasma mass spectroscopy (ICP-MS).
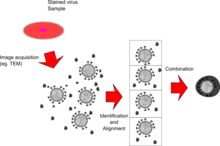
Techniques
Single particle analysis can be done on both negatively stained and vitreous ice-embedded transmission electron cryomicroscopy (CryoTEM) samples. Single particle analysis methods are, in general, reliant on the sample being homogeneous, although techniques for dealing with conformational heterogeneity are being developed.
Images (micrographs) are taken with an electron microscope using charged-coupled device (CCD) detectors coupled to a phosphorescent layer (in the past, they were instead collected on film and digitized using high-quality scanners). The image processing is carried out using specialized software programs, often run on multi-processor computer clusters. Depending on the sample or the desired results, various steps of two- or three-dimensional processing can be done.
In addition, single particle analysis can also be performed in an individual particle mode using an ICP-MS unit.
Alignment and classification
Biological samples, and especially samples embedded in thin vitreous ice, are highly radiation sensitive, thus only low electron doses can be used to image the sample. This low dose, as well as variations in the metal stain used (if used) means images have high noise relative to the signal given by the particle being observed. By aligning several similar images to each other so they are in register and then averaging them, an image with higher signal-to-noise ratio can be obtained. As the noise is mostly randomly distributed and the underlying image features constant, by averaging the intensity of each pixel over several images only the constant features are reinforced. Typically, the optimal alignment (a translation and an in-plane rotation) to map one image onto another is calculated by cross-correlation.
However, a micrograph often contains particles in multiple different orientations and/or conformations, and so to get more representative image averages, a method is required to group similar particle images together into multiple sets. This is normally carried out using one of several data analysis and image classification algorithms, such as multi-variate statistical analysis and hierarchical ascendant classification, or k-means clustering.
Often data sets of tens of thousands of particle images are used, and to reach an optimal solution an iterative procedure of alignment and classification is used, whereby strong image averages produced by classification are used as reference images for a subsequent alignment of the whole data set.
Image filtering
Image filtering (band-pass filtering) is often used to reduce the influence of high and/or low spatial frequency information in the images, which can affect the results of the alignment and classification procedures. This is particularly useful in negative stain images. The algorithms make use of fast Fourier transforms (FFT), often employing Gaussian shaped soft-edged masks in reciprocal space to suppress certain frequency ranges. High-pass filters remove low spatial frequencies (such as ramp or gradient effects), leaving the higher frequencies intact. Low-pass filters remove high spatial frequency features and have a blurring effect on fine details.
Contrast transfer function
Due to the nature of image formation in the electron microscope, bright-field TEM images are obtained using significant underfocus. This, along with features inherent in the microscope's lens system, creates blurring of the collected images visible as a point spread function. The combined effects of the imaging conditions are known as the contrast transfer function (CTF), and can be approximated mathematically as a function in reciprocal space. Specialized image processing techniques such as phase flipping and amplitude correction / Wiener filtering can (at least partially[5]) correct for the CTF, and allow high resolution reconstructions.
Three-dimensional reconstruction
Transmission electron microscopy images are projections of the object showing the distribution of density through the object, similar to medical X-rays. By making use of the projection-slice theorem a three-dimensional reconstruction of the object can be generated by combining many images (2D projections) of the object taken from a range of viewing angles. Proteins in vitreous ice ideally adopt a random distribution of orientations (or viewing angles), allowing a fairly isotropic reconstruction if a large number of particle images are used. This contrasts with electron tomography, where the viewing angles are limited due to the geometry of the sample/imaging set up, giving an anisotropic reconstruction. Filtered back projection is a commonly used method of generating 3D reconstructions in single particle analysis, although many alternative algorithms exist.[3]
Before a reconstruction can be made, the orientation of the object in each image needs to be estimated. Several methods have been developed to work out the relative Euler angles of each image. Some are based on common lines (common 1D projections and sinograms), others use iterative projection matching algorithms. The latter works by beginning with a simple, low resolution 3D starting model and compares the experimental images to projections of the model and creates a new 3D to bootstrap towards a solution.
Methods are also available for making 3D reconstructions of helical samples (such as tobacco mosaic virus), taking advantage of the inherent helical symmetry. Both real space methods (treating sections of the helix as single particles) and reciprocal space methods (using diffraction patterns) can be used for these samples.
Tilt methods
The specimen stage of the microscope can be tilted (typically along a single axis), allowing the single particle technique known as random conical tilt.[6] An area of the specimen is imaged at both zero and at high angle (~60-70 degrees) tilts, or in the case of the related method of orthogonal tilt reconstruction, +45 and −45 degrees. Pairs of particles corresponding to the same object at two different tilts (tilt pairs) are selected, and by following the parameters used in subsequent alignment and classification steps a three-dimensional reconstruction can be generated relatively easily. This is because the viewing angle (defined as three Euler angles) of each particle is known from the tilt geometry.
3D reconstructions from random conical tilt suffer from missing information resulting from a restricted range of orientations. Known as the missing cone (due to the shape in reciprocal space), this causes distortions in the 3D maps. However, the missing cone problem can often be overcome by combining several tilt reconstructions. Tilt methods are best suited to negatively stained samples, and can be used for particles that adsorb to the carbon support film in preferred orientations. The phenomenon known as charging or beam-induced movement makes collecting high-tilt images of samples in vitreous ice challenging.
Map visualization and fitting
Various software programs are available that allow viewing the 3D maps. These often enable the user to manually dock in protein coordinates (structures from X-ray crystallography or NMR) of subunits into the electron density. Several programs can also fit subunits computationally.
Single particle ICP-MS
Single particle-induced coupled plasma-mass spectroscopy (SP-ICP-MS) is used in several areas where there is the possibility of detecting and quantifying suspended particles in samples of environmental fluids, assessing their migration, assessing the size of particles and their distribution, and also determining their stability in a given environment. SP-ICP-MS was designed for particle suspensions in 2000 by Claude Degueldre. He first tested this new methodology at the Forel Institute of the University of Geneva and presented this new analytical approach at the 'Colloid 2oo2' symposium during the spring 2002 meeting of the EMRS, and in the proceedings in 2003.[7] This study presents the theory of SP ICP-MS and the results of tests carried out on clay particles (montmorillonite) as well as other suspensions of colloids. This method was then tested on thorium dioxide nanoparticles by Degueldre & Favarger (2004),[8] zirconium dioxide by Degueldre et al (2004)[9] and gold nanoparticles, which are used as a substrate in nanopharmacy, and published by Degueldre et al (2006).[10] Subsequently, the study of uranium dioxide nano- and micro-particles gave rise to a detailed publication, Ref. Degueldre et al (2006).[11] Since 2010 the interest for SP ICP-MS has exploded.
Examples
- Important information on protein synthesis, ligand binding and RNA interaction can be obtained using this novel technique at medium resolutions of 7.5 to 25Å.[12]
- Methanococcus maripaludis chaperonin,[13] reconstructed to 0.43 nanometer resolution.[14] This bacterial protein complex is a machine for folding other proteins, which get trapped within the shell.
- Fatty acid synthase[15] from yeast at 0.59 nanometer resolution.[16] This huge enzyme complex is responsible for building the long chain fatty acids essential for cellular life.
- A 0.33 nanometer reconstruction of Aquareovirus.[17][18] These viruses infect fish and other aquatic animals. The reconstruction has high enough resolution to have amino acid side chain densities easily visible.
Primary database
References
- Frank, Joachim (2006). Three-dimensional electron microscopy of macromolecular assemblies: visualization of biological molecules in their native state. Oxford: Oxford University Press. ISBN 978-0-19-518218-7.
- Zhou ZH (April 2008). "Towards atomic resolution structural determination by single-particle cryo-electron microscopy". Current Opinion in Structural Biology. 18 (2): 218–28. doi:10.1016/j.sbi.2008.03.004. PMC 2714865. PMID 18403197.
- Wang Q, Matsui T, Domitrovic T, Zheng Y, Doerschuk PC, Johnson JE (March 2013). "Dynamics in cryo EM reconstructions visualized with maximum-likelihood derived variance maps". Journal of Structural Biology. 181 (3): 195–206. doi:10.1016/j.jsb.2012.11.005. PMC 3870017. PMID 23246781.
- Bartesaghi, Alberto; Merk, Alan; Banerjee, Soojay; Matthies, Doreen; Wu, Xiongwu; Milne, Jacqueline L. S.; Subramaniam, Sriram (2015-06-05). "2.2 Å resolution CryoTEM structure of β-galactosidase in complex with a cell-permeant inhibitor". Science. 348 (6239): 1147–1151. Bibcode:2015Sci...348.1147B. doi:10.1126/science.aab1576. ISSN 1095-9203. PMC 6512338. PMID 25953817.
- Downing KH, Glaeser RM (August 2008). "Restoration of weak phase-contrast images recorded with a high degree of defocus: the "twin image" problem associated with CTF correction". Ultramicroscopy. 108 (9): 921–8. doi:10.1016/j.ultramic.2008.03.004. PMC 2694513. PMID 18508199.
- Radermacher M, Wagenknecht T, Verschoor A, Frank J (May 1987). "Three-dimensional reconstruction from a single-exposure, random conical tilt series applied to the 50S ribosomal subunit of Escherichia coli". Journal of Microscopy. 146 (Pt 2): 113–36. doi:10.1111/j.1365-2818.1987.tb01333.x. PMID 3302267.
- C. Degueldre & P. -Y. Favarger, « Colloid analysis by single particle inductively coupled plasma-mass spectroscopy: a feasibility study », Colloids and Surfaces A: Physicochemical and Engineering Aspects, symposium C of the E-MRS 2002 Spring Meeting in Strasbourg, France, vol. 217, no 1, 28 avril 2003, p. 137–142 (ISSN 0927-7757, DOI 10.1016/S0927-7757(02)00568-X)
- C Degueldre et P. -Y Favarger, « Thorium colloid analysis by single particle inductively coupled plasma-mass spectrometry », Talanta, vol. 62, no 5, 19 avril 2004, p. 1051–1054 (ISSN 0039-9140, DOI 10.1016/j.talanta.2003.10.016
- C. Degueldre, P. -Y. Favarger et C. Bitea, « Zirconia colloid analysis by single particle inductively coupled plasma–mass spectrometry », Analytica Chimica Acta, vol. 518, no 1, 2 août 2004, p. 137–142 (ISSN 0003-2670, DOI 10.1016/j.aca.2004.04.015)
- C. Degueldre, P. -Y. Favarger et S. Wold, « Gold colloid analysis by inductively coupled plasma-mass spectrometry in a single particle mode », Analytica Chimica Acta, vol. 555, no 2, 12 janvier 2006, p. 263–268 (ISSN 0003-2670, DOI 10.1016/j.aca.2005.09.021)
- C. Degueldre, P. -Y. Favarger, R. Rossé et S. Wold, « Uranium colloid analysis by single particle inductively coupled plasma-mass spectrometry », Talanta, vol. 68, no 3, 15 janvier 2006, p. 623–628 (ISSN 0039-9140, DOI 10.1016/j.talanta.2005.05.006,
- Arias-Palomo E, Recuero-Checa MA, Bustelo XR, Llorca O (December 2007). "3D structure of Syk kinase determined by single-particle electron microscopy". Biochim. Biophys. Acta. 1774 (12): 1493–9. doi:10.1016/j.bbapap.2007.10.008. PMC 2186377. PMID 18021750.
- Japanese Protein databank http://www.pdbj.org/emnavi/emnavi_movie.php?id=5137
- Zhang J, Baker ML, Schröder GF, et al. (January 2010). "Mechanism of folding chamber closure in a group II chaperonin". Nature. 463 (7279): 379–83. Bibcode:2010Natur.463..379Z. doi:10.1038/nature08701. PMC 2834796. PMID 20090755.
- Japanese Protein databank http://www.pdbj.org/emnavi/emnavi_movie.php?id=1623
- Gipson P, Mills DJ, Wouts R, Grininger M, Vonck J, Kühlbrandt W (May 2010). "Direct structural insight into the substrate-shuttling mechanism of yeast fatty acid synthase by electron cryomicroscopy". Proceedings of the National Academy of Sciences of the United States of America. 107 (20): 9164–9. Bibcode:2010PNAS..107.9164G. doi:10.1073/pnas.0913547107. PMC 2889056. PMID 20231485.
- Japanese Protein databank http://www.pdbj.org/emnavi/emnavi_movie.php?id=5160
- Zhang X, Jin L, Fang Q, Hui WH, Zhou ZH (April 2010). "3.3 A cryo-EM structure of a nonenveloped virus reveals a priming mechanism for cell entry". Cell. 141 (3): 472–82. doi:10.1016/j.cell.2010.03.041. PMC 3422562. PMID 20398923.
External links
| Library resources about Single particle analysis |