Liquid–liquid extraction
Liquid–liquid extraction (LLE), also known as solvent extraction and partitioning, is a method to separate compounds or metal complexes, based on their relative solubilities in two different immiscible liquids, usually water (polar) and an organic solvent (non-polar). There is a net transfer of one or more species from one liquid into another liquid phase, generally from aqueous to organic. The transfer is driven by chemical potential, i.e. once the transfer is complete, the overall system of chemical components that make up the solutes and the solvents are in a more stable configuration (lower free energy). The solvent that is enriched in solute(s) is called extract. The feed solution that is depleted in solute(s) is called the raffinate. LLE is a basic technique in chemical laboratories, where it is performed using a variety of apparatus, from separatory funnels to countercurrent distribution equipment called as mixer settlers. This type of process is commonly performed after a chemical reaction as part of the work-up, often including an acidic work-up.
The term partitioning is commonly used to refer to the underlying chemical and physical processes involved in liquid–liquid extraction, but on another reading may be fully synonymous with it. The term solvent extraction can also refer to the separation of a substance from a mixture by preferentially dissolving that substance in a suitable solvent. In that case, a soluble compound is separated from an insoluble compound or a complex matrix.
From a hydrometallurgical perspective, solvent extraction is exclusively used in separation and purification of uranium and plutonium, zirconium and hafnium, separation of cobalt and nickel, separation and purification of rare earth elements etc., its greatest advantage being its ability to selectively separate out even very similar metals. One obtains high-purity single metal streams on 'stripping' out the metal value from the 'loaded' organic wherein one can precipitate or deposit the metal value. Stripping is the opposite of extraction: Transfer of mass from organic to aqueous phase.
LLE is also widely used in the production of fine organic compounds, the processing of perfumes, the production of vegetable oils and biodiesel, and other industries. It is among the most common initial separation techniques, though some difficulties result in extracting out closely related functional groups.
Liquid–liquid extraction is possible in non-aqueous systems: In a system consisting of a molten metal in contact with molten salts, metals can be extracted from one phase to the other. This is related to a mercury electrode where a metal can be reduced, the metal will often then dissolve in the mercury to form an amalgam that modifies its electrochemistry greatly. For example, it is possible for sodium cations to be reduced at a mercury cathode to form sodium amalgam, while at an inert electrode (such as platinum) the sodium cations are not reduced. Instead, water is reduced to hydrogen. A detergent or fine solid can be used to stabilize an emulsion, or third phase.
Measures of effectiveness
Distribution ratio
In solvent extraction, a distribution ratio is often quoted as a measure of how well-extracted a species is. The distribution ratio (Kd) is equal to the concentration of a solute in the organic phase divided by its concentration in the aqueous phase. Depending on the system, the distribution ratio can be a function of temperature, the concentration of chemical species in the system, and a large number of other parameters. Note that D is related to the ΔG of the extraction process.
Sometimes, the distribution ratio is referred to as the partition coefficient, which is often expressed as the logarithm. Note that a distribution ratio for uranium and neptunium between two inorganic solids (zirconolite and perovskite) has been reported.[1] In solvent extraction, two immiscible liquids are shaken together. The more polar solutes dissolve preferentially in the more polar solvent, and the less polar solutes in the less polar solvent. In this experiment, the nonpolar halogens preferentially dissolve in the non-polar mineral oil.[2]
Although the distribution ratio and partition coefficient are often used synonymously, they are not necessarily so. Solutes may exist in more than one form in any particular phase, which would mean that the partition coefficient (Kd) and distribution ratio (D) will have different values. This is an important distinction to make as whilst the partition coefficient has a fixed value for the partitioning of a solute between two phases, the distribution ratio changes with differing conditions in the solvent.[3]
After performing liquid–liquid extraction, a quantitative measure must be taken to determine the ratio of the solution's total concentration in each phase of the extraction. This quantitative measure is known as the distribution ratio or distribution coefficient.[4]
Separation factors
The separation factor is one distribution ratio divided by another; it is a measure of the ability of the system to separate two solutes. For instance, if the distribution ratio for nickel (DNi) is 10 and the distribution ratio for silver (DAg) is 100, then the silver/nickel separation factor (SFAg/Ni) is equal to DAg/DNi = SFAg/Ni = 10.[5]
Decontamination factor
This is used to express the ability of a process to remove a contaminant from a product. For instance, if a process is fed with a mixture of 1:9 cadmium to indium, and the product is a 1:99 mixture of cadmium and indium, then the decontamination factor (for the removal of cadmium) of the process is 0.11 / 0.01 = 11.
Slopes of graphs
The easy way to work out the extraction mechanism is to draw graphs and measure the slopes. If for an extraction system the D value is proportional to the square of the concentration of a reagent (Z) then the slope of the graph of log10(D) against log10([[Z]]) will be two.
Measures of success
Success of liquid–liquid extraction is measured through separation factors and decontamination factors. The best way to understand the success of an extraction column is through the liquid–liquid equilibrium (LLE) data set. The data set can then be converted into a curve to determine the steady state partitioning behavior of the solute between the two phases. The y-axis is the concentration of solute in the extract (solvent) phase, and the x-axis is the concentration of the solute in the raffinate phase. From here, one can determine steps for optimization of the process.[6]
Techniques
Batchwise single stage extractions
This is commonly used on the small scale in chemical labs. It is normal to use a separating funnel. Processes include DLLME and direct organic extraction. After equilibration, the extract phase containing the desired solute is separated out for further processing.
Dispersive liquid–liquid microextraction (DLLME)
A process used to extract small amounts of organic compounds from water samples.[7] This process is done by injecting small amounts of an appropriate extraction solvent (C2Cl4) and a disperser solvent (acetone) into the aqueous solution. The resulting solution is then centrifuged to separate the organic and aqueous layers. This process is useful in extraction organic compounds such as organochloride and organophsophorus pesticides, as well as substituted benzene compounds from water samples.[7]
Direct organic extraction
By mixing partially organic soluble samples in organic solvent (toluene, benzene, xylene), the organic soluble compounds will dissolve into the solvent and can be separated using a separatory funnel. This process is valuable in the extraction of proteins and specifically phosphoprotein and phosphopeptide phosphatases.[8]
Another example of this application is extracting anisole from a mixture of water and 5% acetic acid using ether, then the anisole will enter the organic phase. The two phases would then be separated. The acetic acid can then be scrubbed (removed) from the organic phase by shaking the organic extract with sodium bicarbonate. The acetic acid reacts with the sodium bicarbonate to form sodium acetate, carbon dioxide, and water.
Caffeine can also be extracted from coffee beans and tea leaves using a direct organic extraction. The beans or leaves can be soaked in ethyl acetate which favorably dissolves the caffeine, leaving a majority of the coffee or tea flavor remaining in the initial sample.[9]
Multistage countercurrent continuous processes
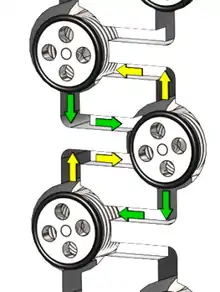
These are commonly used in industry for the processing of metals such as the lanthanides; because the separation factors between the lanthanides are so small many extraction stages are needed.[10] In the multistage processes, the aqueous raffinate from one extraction unit is fed to the next unit as the aqueous feed, while the organic phase is moved in the opposite direction. Hence, in this way, even if the separation between two metals in each stage is small, the overall system can have a higher decontamination factor.
Multistage countercurrent arrays have been used for the separation of lanthanides. For the design of a good process, the distribution ratio should be not too high (>100) or too low (<0.1) in the extraction portion of the process. It is often the case that the process will have a section for scrubbing unwanted metals from the organic phase, and finally a stripping section to obtain the metal back from the organic phase.
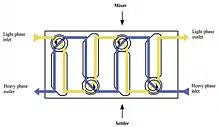
Mixer–settlers
Battery of mixer-settlers counter currently interconnected. Each mixer-settler unit provides a single stage of extraction. A mixer settler consists of a first stage that mixes the phases together followed by a quiescent settling stage that allows the phases to separate by gravity.

A novel settling device, Sudhin BioSettler, can separate an oil-water emulsion continuously at a much faster rate than simple gravity settlers. In this photo, an oil-water emulsion, stirred by an impeller in an external reservoir and pumped continuously into the two bottom side ports of BioSettler, is separated very quickly into a clear organic (mineral oil) layer exiting via the top of BioSettler and an aqueous (coloured with a red food dye) layer being pumped out continuously from the bottom of BioSettler.
In the multistage countercurrent process, multiple mixer settlers are installed with mixing and settling chambers located at alternating ends for each stage (since the outlet of the settling sections feed the inlets of the adjacent stage's mixing sections). Mixer-settlers are used when a process requires longer residence times and when the solutions are easily separated by gravity. They require a large facility footprint, but do not require much headspace, and need limited remote maintenance capability for occasional replacement of mixing motors. (Colven, 1956; Davidson, 1957)[11]
Centrifugal extractors
Centrifugal extractors mix and separate in one unit. Two liquids will be intensively mixed between the spinning rotor and the stationary housing at speeds up to 6000 RPM. This develops great surfaces for an ideal mass transfer from the aqueous phase into the organic phase. At 200–2000 g, both phases will be separated again. Centrifugal extractors minimize the solvent in the process, optimize the product load in the solvent and extract the aqueous phase completely. Counter current and cross current extractions are easily established.[12]
Extraction without chemical change
Some solutes such as noble gases can be extracted from one phase to another without the need for a chemical reaction (see absorption). This is the simplest type of solvent extraction. When a solvent is extracted, two immiscible liquids are shaken together. The more polar solutes dissolve preferentially in the more polar solvent, and the less polar solutes in the less polar solvent. Some solutes that do not at first sight appear to undergo a reaction during the extraction process do not have distribution ratio that is independent of concentration. A classic example is the extraction of carboxylic acids (HA) into nonpolar media such as benzene. Here, it is often the case that the carboxylic acid will form a dimer in the organic layer so the distribution ratio will change as a function of the acid concentration (measured in either phase).
For this case, the extraction constant k is described by k = [HAorganic]2/[HAaqueous]
Solvation mechanism
Using solvent extraction it is possible to extract uranium, plutonium, thorium and many rare earth elements from acid solutions in a selective way by using the right choice of organic extracting solvent and diluent. One solvent used for this purpose is the organophosphate tributyl phosphate (TBP). The PUREX process that is commonly used in nuclear reprocessing uses a mixture of tri-n-butyl phosphate and an inert hydrocarbon (kerosene), the uranium(VI) are extracted from strong nitric acid and are back-extracted (stripped) using weak nitric acid. An organic soluble uranium complex [UO2(TBP)2(NO3)2] is formed, then the organic layer bearing the uranium is brought into contact with a dilute nitric acid solution; the equilibrium is shifted away from the organic soluble uranium complex and towards the free TBP and uranyl nitrate in dilute nitric acid. The plutonium(IV) forms a similar complex to the uranium(VI), but it is possible to strip the plutonium in more than one way; a reducing agent that converts the plutonium to the trivalent oxidation state can be added. This oxidation state does not form a stable complex with TBP and nitrate unless the nitrate concentration is very high (circa 10 mol/L nitrate is required in the aqueous phase). Another method is to simply use dilute nitric acid as a stripping agent for the plutonium. This PUREX chemistry is a classic example of a solvation extraction. In this case, DU = k [TBP]2[NO3-]2.
Ion exchange mechanism
Another extraction mechanism is known as the ion exchange mechanism. Here, when an ion is transferred from the aqueous phase to the organic phase, another ion is transferred in the other direction to maintain the charge balance. This additional ion is often a hydrogen ion; for ion exchange mechanisms, the distribution ratio is often a function of pH. An example of an ion exchange extraction would be the extraction of americium by a combination of terpyridine and a carboxylic acid in tert-butyl benzene. In this case
- DAm = k [terpyridine]1[carboxylic acid]3[H+]−3
Another example is the extraction of zinc, cadmium, or lead by a dialkyl phosphinic acid (R2PO2H) into a nonpolar diluent such as an alkane. A non-polar diluent favours the formation of uncharged non-polar metal complexes.
Some extraction systems are able to extract metals by both the solvation and ion exchange mechanisms; an example of such a system is the americium (and lanthanide) extraction from nitric acid by a combination of 6,6'-bis-(5,6-dipentyl-1,2,4-triazin-3-yl)-2,2'-bipyridine and 2-bromohexanoic acid in tert-butyl benzene. At both high- and low-nitric acid concentrations, the metal distribution ratio is higher than it is for an intermediate nitric acid concentration.
Ion pair extraction
It is possible by careful choice of counterion to extract a metal. For instance, if the nitrate concentration is high, it is possible to extract americium as an anionic nitrate complex if the mixture contains a lipophilic quaternary ammonium salt.
An example that is more likely to be encountered by the 'average' chemist is the use of a phase transfer catalyst. This is a charged species that transfers another ion to the organic phase. The ion reacts and then forms another ion, which is then transferred back to the aqueous phase.
For instance, the 31.1 kJ mol−1 is required to transfer an acetate anion into nitrobenzene,[13] while the energy required to transfer a chloride anion from an aqueous phase to nitrobenzene is 43.8 kJ mol−1.[14] Hence, if the aqueous phase in a reaction is a solution of sodium acetate while the organic phase is a nitrobenzene solution of benzyl chloride, then, when a phase transfer catalyst, the acetate anions can be transferred from the aqueous layer where they react with the benzyl chloride to form benzyl acetate and a chloride anion. The chloride anion is then transferred to the aqueous phase. The transfer energies of the anions contribute to that given out by the reaction.
A 43.8 to 31.1 kJ mol−1 = 12.7 kJ mol−1 of additional energy is given out by the reaction when compared with energy if the reaction had been done in nitrobenzene using one equivalent weight of a tetraalkylammonium acetate.[15]
Types of aqueous two-phase extractions
Polymer–polymer systems. In a Polymer–polymer system, both phases are generated by a dissolved polymer. The heavy phase will generally be a polysaccharide, and the light phase is generally Polyethylene glycol (PEG). Traditionally, the polysaccharide used is dextran. However, dextran is relatively expensive, and research has been exploring using less expensive polysaccharides to generate the heavy phase. If the target compound being separated is a protein or enzyme, it is possible to incorporate a ligand to the target into one of the polymer phases. This improves the target's affinity to that phase, and improves its ability to partition from one phase into the other. This, as well as the absence of solvents or other denaturing agents, makes polymer–polymer extractions an attractive option for purifying proteins. The two phases of a polymer–polymer system often have very similar densities, and very low surface tension between them. Because of this, demixing a polymer–polymer system is often much more difficult than demixing a solvent extraction. Methods to improve the demixing include centrifugation, and application of an electric field.
Polymer–salt systems. Aqueous two-phase systems can also be generated by generating the heavy phase with a concentrated salt solution. The polymer phase used is generally still PEG. Generally, a kosmotropic salt, such as Na3PO4 is used, however PEG–NaCl systems have been documented when the salt concentration is high enough. Since polymer–salt systems demix readily they are easier to use. However, at high salt concentrations, proteins generally either denature, or precipitate from solution. Thus, polymer–salt systems are not as useful for purifying proteins.
Ionic liquids systems. Ionic liquids are ionic compounds with low melting points. While they are not technically aqueous, recent research has experimented with using them in an extraction that does not use organic solvents.
DNA purification
The ability to purify DNA from a sample is important for many modern biotechnology processes. However, samples often contain nucleases that degrade the target DNA before it can be purified. It has been shown that DNA fragments will partition into the light phase of a polymer–salt separation system. If ligands known to bind and deactivate nucleases are incorporated into the polymer phase, the nucleases will then partition into the heavy phase and be deactivated. Thus, this polymer–salt system is a useful tool for purifying DNA from a sample while simultaneously protecting it from nucleases.
Food industry
The PEG–NaCl system has been shown to be effective at partitioning small molecules, such as peptides and nucleic acids. These compounds are often flavorants or odorants. The system could then be used by the food industry to isolate or eliminate particular flavors. Caffeine extraction used to be done using liquid–liquid extraction, specifically direct and indirect liquid–liquid extraction (Swiss Water Method), but has since moved towards super-critical CO2 as it is cheaper and can be done on a commercial scale.[16][17]
Analytical chemistry
Often there are chemical species present or necessary at one stage of sample processing that will interfere with the analysis. For example, some air monitoring is performed by drawing air through a small glass tube filled with sorbent particles that have been coated with a chemical to stabilize or derivatize the analyte of interest. The coating may be of such a concentration or characteristics that it would damage the instrumentation or interfere with the analysis. If the sample can be extracted from the sorbent using a nonpolar solvent (such as toluene or carbon disulfide), and the coating is polar (such as HBr or phosphoric acid) the dissolved coating will partition into the aqueous phase. Clearly the reverse is true as well, using polar extraction solvent and a nonpolar solvent to partition a nonpolar interferent. A small aliquot of the organic phase (or in the latter case, polar phase) can then be injected into the instrument for analysis.
Purification of amines
Amines (analogously to ammonia) have a lone pair of electrons on the nitrogen atom that can form a relatively weak bond to a hydrogen atom. It is therefore the case that under acidic conditions amines are typically protonated, carrying a positive charge and under basic conditions they are typically deprotonated and neutral. Amines of sufficiently low molecular weight are rather polar and can form hydrogen bonds with water and therefore will readily dissolve in aqueous solutions. Deprotonated amines on the other hand, are neutral and have greasy, nonpolar organic substituents, and therefore have a higher affinity for nonpolar inorganic solvents. As such purification steps can be carried out where an aqueous solution of an amine is neutralized with a base such as sodium hydroxide, then shaken in a separatory funnel with a nonpolar solvent that is immiscible with water. The organic phase is then drained off. Subsequent processing can recover the amine by techniques such as recrystallization, evaporation or distillation; subsequent extraction back to a polar phase can be performed by adding HCl and shaking again in a separatory funnel (at which point the ammonium ion could be recovered by adding an insoluble counterion), or in either phase, reactions could be performed as part of a chemical synthesis.
Temperature swing solvent extraction
Temperature swing solvent extraction is an experimental technique for the desalination of drinking water. It has been used to remove up to 98.4% of the salt content in water, and is able to process hypersaline brines that cannot be desalinated using reverse osmosis.[18]
Kinetics of extraction
It is important to investigate the rate at which the solute is transferred between the two phases, in some cases by an alteration of the contact time it is possible to alter the selectivity of the extraction. For instance, the extraction of palladium or nickel can be very slow because the rate of ligand exchange at these metal centers is much lower than the rates for iron or silver complexes.
Aqueous complexing agents
If a complexing agent is present in the aqueous phase then it can lower the distribution ratio. For instance, in the case of iodine being distributed between water and an inert organic solvent such as carbon tetrachloride then the presence of iodide in the aqueous phase can alter the extraction chemistry: instead of being a constant it becomes

- = k[I2 (organic)]/[I2 (aq)][I− (aq)]
This is because the iodine reacts with the iodide to form I3−. The I3− anion is an example of a polyhalide anion that is quite common.
Industrial process design
In a typical scenario, an industrial process will use an extraction step in which solutes are transferred from the aqueous phase to the organic phase; this is often followed by a scrubbing stage in which unwanted solutes are removed from the organic phase, then a stripping stage in which the wanted solutes are removed from the organic phase. The organic phase may then be treated to make it ready for use again.[19][20]
After use, the organic phase may be subjected to a cleaning step to remove any degradation products; for instance, in PUREX plants, the used organic phase is washed with sodium carbonate solution to remove any dibutyl hydrogen phosphate or butyl dihydrogen phosphate that might be present.
Liquid-liquid equilibrium calculations
In order to calculate the phase equilibrium, it is necessary to use a thermodynamic model such as NRTL, UNIQUAC, etc. The corresponding parameters of these models can be obtained from literature (e.g. Dechema Chemistry Data Series, Dortmund Data Bank, etc.) or by a correlation process of experimental data.[21][22][23][24]
Equipment
While solvent extraction is often done on a small scale by synthetic lab chemists using a separatory funnel, Craig apparatus or membrane-based techniques,[25] it is normally done on the industrial scale using machines that bring the two liquid phases into contact with each other. Such machines include centrifugal contactors, Thin Layer Extraction, spray columns, pulsed columns, and mixer-settlers.
Extraction of metals
The extraction methods for a range of metals include:[26][27]
Cobalt
The extraction of cobalt from hydrochloric acid using Alamine 336 (tri-octyl/decyl amine) in meta-xylene.[28] Cobalt can be extracted also using Ionquest 290 or Cyanex 272 {bis-(2,4,4-trimethylpentyl) phosphinic acid}.
Copper
Copper can be extracted using hydroxyoximes as extractants, a recent paper describes an extractant that has a good selectivity for copper over cobalt and nickel.[29]
Neodymium
The rare earth element Neodymium is extracted by di(2-ethyl-hexyl)phosphoric acid into hexane by an ion exchange mechanism.[30]
Nickel
Nickel can be extracted using di(2-ethyl-hexyl)phosphoric acid and tributyl phosphate in a hydrocarbon diluent (Shellsol).[31]
Palladium and platinum
Dialkyl sulfides, tributyl phosphate and alkyl amines have been used for extracting palladium and platinum.[32][33]
Polonium
Polonium is produced in reactors from natural 209Bi, bombarded with neutrons, creating 210Bi, which then decays to 210Po via beta-minus decay. The final purification is done pyrochemically followed by liquid-liquid extraction vs sodium hydroxide at 500 deg C.[34]
Zinc and cadmium
Zinc and cadmium are both extracted by an ion exchange process, the N,N,N′,N′-tetrakis(2-pyridylmethyl)ethylenediamine (TPEN) acts as a masking agent for the zinc and an extractant for the cadmium.[35] In the modified Zincex process, zinc is separated from most divalent ions by solvent extraction. D2EHPA (Di (2) ethyl hexyl phosphoric acid) is used for this. A zinc ion replaces the proton from two D2EHPA molecules. To strip the zinc from the D2EHPA, sulfuric acid is used, at a concentration of above 170g/L (typically 240-265g/L).
Lithium
Lithium extraction is more popular due to the high demand of lithium-ion batteries. TBP (Tri-butyl phosphate) and FeCl3 are mostly used to extract lithium from brine (with high Li/Mg ratio).[36] Alternatively, Cyanex 272 was also used to extract lithium. The mechanism of lithium extraction was found differently from other metals, such as cobalt, due to the weak coordinating bonding between lithium ions and extractants.[37]
See also
- Fragrance extraction
- Dortmund Data Bank
- Non-random two-liquid model - (NRTL model) LL Phase Equilibrium Calculation
- UNIQUAC - LL Phase Equilibrium Calculation
References
- "SSRL Publications & Reports | Stanford Synchrotron Radiation Lightsource" (PDF). Archived from the original (PDF) on March 9, 2008. Retrieved January 21, 2006.
- pnjjrose. "Solvent Extraction Notes".
- "7.7: Liquid–Liquid Extractions". Chemistry LibreTexts. 2013-10-25. Retrieved 2017-03-26.
- http://courses.chem.psu.edu/chem36/Experiments/PDF's_for_techniques/Liquid_Liquid.pdf
- "Basic Technology and Tools in Chemical Engineering Field - S. Wesley - Documents".
- "Archived copy" (PDF). Archived from the original (PDF) on 2015-09-29. Retrieved 2015-09-28.
{{cite web}}: CS1 maint: archived copy as title (link) - Rezaee, Mohammad; Assadi, Yaghoub; Milani Hosseini, Mohammad-Reza; Aghaee, Elham; Ahmadi, Fardin; Berijani, Sana (2006). "Determination of organic compounds in water using dispersive liquid–liquid microextraction". Journal of Chromatography A. 1116 (1–2): 1–9. doi:10.1016/j.chroma.2006.03.007. ISSN 0021-9673. PMID 16574135.
- Shacter, Emily (1984). "Organic extraction of Pi with isobutanol/toluene". Analytical Biochemistry. 138 (2): 416–420. doi:10.1016/0003-2697(84)90831-5. ISSN 0003-2697. PMID 6742419.
- Senese F (20 September 2005). "How is coffee decaffeinated?". General Chemistry Online. Retrieved 3 August 2009.
- Binnemans, Koen (2007). "Lanthanides and Actinides in Ionic Liquids". Chemical Reviews. 107 (6): 2592–2614. doi:10.1021/cr050979c. ISSN 0009-2665. PMID 17518503.
- Liquid–Liquid Extraction Equipment, Jack D. Law and Terry A. Todd, Idaho National Laboratory.
- James A. Kent, ed. (2003). Riegel's Handbook of Industrial Chemistry (10th ed.). Springer. doi:10.1007/0-387-23816-6. ISBN 978-0-306-47411-8.
- Scholz, F.; S. Komorsky-Lovric; M. Lovric (February 2000). "A new access to Gibbs energies of transfer of ions across liquid|liquid interfaces and a new method to study electrochemical processes at well-defined three-phase junctions". Electrochemistry Communications. 2 (2): 112–118. doi:10.1016/S1388-2481(99)00156-3.
- de Namor, Angela F. Danil; Hill, Tony; Sigstad, Elizabeth (1983). "Free energies of transfer of 1: 1 electrolytes from water to nitrobenzene. Partition of ions in the water + nitrobenzene system". Journal of the Chemical Society, Faraday Transactions 1: Physical Chemistry in Condensed Phases. 79 (11): 2713. doi:10.1039/f19837902713. ISSN 0300-9599.
- zamani, Dariush. "Extraction Operation".
- Peker, Hulya; Srinivasan, M. P.; Smith, J. M.; McCoy, Ben J. (1992). "Caffeine extraction rates from coffee beans with supercritical carbon dioxide". AIChE Journal. 38 (5): 761–770. doi:10.1002/aic.690380513. ISSN 0001-1541.
- Emden, Lorenzo (6 July 2012). "Decaffeination 101: Four Ways to Decaffeinate Coffee". Coffee Confidential. Retrieved 29 October 2014.
- Evarts, Holly (6 May 2019). "Radical Desalination Approach May Disrupt the Water Industry". Columbia Engineering. Retrieved 24 January 2021.
- Reyes-Labarta, J.A.; Olaya, M.M.; Gómez, A.; Marcilla, A. (1999). "New method for quaternary systems liquid-liquid extraction tray to tray design". Industrial & Engineering Chemistry Research. 38 (8): 3083–3095. doi:10.1021/ie9900723.
- Reyes-Labarta, J.A.; Grossmann, I.E (2001). "Disjunctive Programming Models for the Optimal Design of Liquid-Liquid Multistage Extractors and Separation Sequences". AIChE Journal. 47 (10): 2243–2252. doi:10.1002/aic.690471011.
- Reyes-Labarta, J.A.; Olaya, M.M.; Velasco, R.; Serrano, M.D.; Marcilla, A. (2009). "Correlation of the Liquid-Liquid Equilibrium Data for Specific Ternary Systems with One or Two Partially Miscible Binary Subsystems". Fluid Phase Equilibria. 278 (1–2): 9–14. doi:10.1016/j.fluid.2008.12.002.
- Marcilla, A.; Reyes-Labarta, J.A.; Serrano, M.D.; Olaya, M.M. (2011). "GE Models and Algorithms for Condensed Phase Equilibrium Data Regression in Ternary Systems: Limitations and Proposals". The Open Thermodynamics Journal. 5: 48–62. doi:10.2174/1874396X01105010048.
- Marcilla, Antonio; Reyes-Labarta, Juan A.; Olaya, M.Mar (2017). "Should we trust all the published LLE correlation parameters in phase equilibria? Necessity of their Assessment Prior to Publication". Fluid Phase Equilibria. 433: 243–252. doi:10.1016/j.fluid.2016.11.009. hdl:10045/66521.
- Labarta, Juan A.; Olaya, Maria del Mar; Marcilla, Antonio (2015-11-27). "Graphical User Interface (GUI) for the analysis of Gibbs Energy surfaces, including LL tie-lines and Hessian matrix". University of Alicante. hdl:10045/51725.
- Adamo, Andrea; Heider, Patrick L.; Weeranoppanant, Nopphon; Jensen, Klavs F. (2013). "Membrane-Based, Liquid–Liquid Separator with Integrated Pressure Control" (PDF). Industrial & Engineering Chemistry Research. 52 (31): 10802–10808. doi:10.1021/ie401180t. hdl:1721.1/92770. ISSN 0888-5885.
- Mackenzie, Murdoch. "The Solvent Extraction of Some Major Metals" (PDF). Cognis GmbH. Archived from the original (PDF) on 2010-01-04. Retrieved 2008-11-18.
- Patel, Madhav; Karamalidis, Athanasios K. (May 2021). "Germanium: A review of its US demand, uses, resources, chemistry, and separation technologies". Separation and Purification Technology. 275: 118981. doi:10.1016/j.seppur.2021.118981. ISSN 1383-5866.
- Filiz, M.; Sayar, N.A.; Sayar, A.A. (2006). "Extraction of cobalt(II) from aqueous hydrochloric acid solutions into Alamine 336–m-xylene mixtures". Hydrometallurgy. 81 (3–4): 167–173. Bibcode:2006HydMe..81..167F. doi:10.1016/j.hydromet.2005.12.007. ISSN 0304-386X.
- Baba, Yoshinari; Iwakuma, Minako; Nagami, Hideto (2002). "Extraction Mechanism for Copper(II) with 2-Hydroxy-4-n-octyloxybenzophenone Oxime". Industrial & Engineering Chemistry Research. 41 (23): 5835–5841. doi:10.1021/ie0106736. ISSN 0888-5885.
- Sanchez, J.M.; Hidalgo, M.; Salvadó, V.; Valiente, M. (1999). "Extraction of Neodymium(III) at Trace Level with Di(2-Ethyl-Hexyl)Phosphoric Acid in Hexane". Solvent Extraction and Ion Exchange. 17 (3): 455–474. doi:10.1080/07366299908934623. ISSN 0736-6299.
- Lee W. John. "A Potential Nickel / Cobalt Recovery Process". BioMetallurgical Pty Ltd. Archived from the original on 2008-09-26. Retrieved 2006-03-31.
- "Precious Metals Refining By Solvent Extraction". Halwachs Edelmetallchemie und Verfahrenstechnik. Retrieved 2008-11-18.
- Giridhar, P.; Venkatesan, K.A.; Srinivasan, T.G.; Vasudeva Rao, P.R. (2006). "Extraction of fission palladium by Aliquat 336 and electrochemical studies on direct recovery from ionic liquid phase". Hydrometallurgy. 81 (1): 30–39. Bibcode:2006HydMe..81...30G. doi:10.1016/j.hydromet.2005.10.001. ISSN 0304-386X.
- Schulz, Wallace W.; Schiefelbein, Gary F.; Bruns, Lester E. (1969). "Pyrochemical Extraction of Polonium from Irradiated Bismuth Metal". Ind. Eng. Chem. Process Des. Dev. 8 (4): 508–515. doi:10.1021/i260032a013.
- K. Takeshita; K. Watanabe; Y. Nakano; M. Watanabe (2003). "Solvent extraction separation of Cd(II) and Zn(II) with the organophosphorus extractant D2EHPA and the aqueous nitrogen-donor ligand TPEN". Hydrometallurgy. 70 (1–3): 63–71. Bibcode:2003HydMe..70...63T. doi:10.1016/s0304-386x(03)00046-x.
- Wesselborg, Tobias; Virolainen, Sami; Sainio, Tuomo (2021-06-01). "Recovery of lithium from leach solutions of battery waste using direct solvent extraction with TBP and FeCl3". Hydrometallurgy. 202: 105593. Bibcode:2021HydMe.20205593W. doi:10.1016/j.hydromet.2021.105593. ISSN 0304-386X. S2CID 233662976.
- Lu, Junnan; Stevens, Geoff W.; Mumford, Kathryn A. (2021-12-01). "Development of heterogeneous equilibrium model for lithium solvent extraction using organophosphinic acid". Separation and Purification Technology. 276: 119307. doi:10.1016/j.seppur.2021.119307. ISSN 1383-5866.
Further reading
- B.L. Karger, 2014, "Separation and Purification: Single-stage versus multistage processes" and "Separation and Purification: Separations Based on Equilibrium", Encyclopædia Britannica, see and , accessed 12 May 2014.
- Gunt Hamburg, 2014, "Thermal Process Engineering: liquid–liquid extraction and solid-liquid extraction", see , accessed 12 May 2014.
- G.W. Stevens, T.C., Lo, & M. H. I. Baird, 2007, "Extraction, liquid–liquid", in Kirk-Othmer Encyclopedia of Chemical Technology, doi:10.1002/0471238961.120917211215.a01.pub2, accessed 12 May 2014.
- Colin Poole & Michael Cooke, 2000, "Extraction", in Encyclopedia of Separation Science, 10 Vols., ISBN 978-0-12-226770-3, see , accessed 12 May 2014.
- Sikdar, Cole, et al. Aqueous Two-Phase Extractions in Bioseparations: An Assessment. Biotechnology 9:254. 1991
- Szlag, Giuliano. A Low-Cost Aqueous Two Phase System for Enzyme Extraction. Biotechnology Techniques 2:4:277. 1988
- Dreyer, Kragl. Ionic Liquids for Aqueous Two-Phase Extraction and Stabilization of Enzymes. Biotechnology and Bioengineering. 99:6:1416. 2008
- Boland. Aqueous Two-Phase Systems: Methods and Protocols. Pg 259-269
- https://web.archive.org/web/20100702074135/http://ull.chemistry.uakron.edu/chemsep/extraction/
- Topological Analysis of the Gibbs Energy Function (Liquid-Liquid Equilibrium Correlation Data). Including a Thermodynamic Review and a Graphical User Interface (GUI) for Surfaces/Tie-lines/Hessian matrix analysis - University of Alicante (Reyes-Labarta et al. 2015-18)