Evolution of ageing
Enquiry into the evolution of ageing, or aging, aims to explain why a detrimental process such as ageing would evolve, and why there is so much variability in the lifespans of organisms. The classical theories of evolution (mutation accumulation, antagonistic pleiotropy, and disposable soma)[1][2][3] suggest that environmental factors, such as predation, accidents, disease, and/or starvation, ensure that most organisms living in natural settings will not live until old age, and so there will be very little pressure to conserve genetic changes that increase longevity. Natural selection will instead strongly favor genes which ensure early maturation and rapid reproduction, and the selection for genetic traits which promote molecular and cellular self-maintenance will decline with age for most organisms.[4]
Theories and hypotheses
The beginning
August Weismann was responsible for interpreting and formalizing the mechanisms of Darwinian evolution in a modern theoretical framework. In 1889, he theorized that ageing was part of life's program to make room for the next generation in order to sustain the turnover that is necessary for evolution.[5] The idea that the ageing characteristic was selected (an adaptation) because of its deleterious effect was largely discounted for much of the 20th century, but a theoretical model suggests that altruistic ageing could evolve if there is little migration among populations.[6] Weismann later abandoned his theory and after some time followed up with his "programmed death" theory.
Natural selection is a process that allows organisms to better adapt to the environment, it is the survival of the fittest which are predicted to produce more offsprings. Natural selection acts on life history traits in order to optimize reproductive success and lifetime fitness. Fitness in this context refers to how likely an organism is to survive and reproduce. It is based on the environment and is also relative to other individuals in the population. Examples of life history traits include; age and size at first reproduction, number of size and offsprings produced, and the period of reproductive lifespan. Organisms put energy into growth, reproduction, and maintenance by following a particular pattern which changes throughout their lifetime due to the trade-offs that exist between the different energy allocations. Investment in current vs future reproduction, for example, comes at the expense of the other. Natural selection, however is not so effective on organisms as they age. Mutation accumulation (MA) and antagonistic pleiotropy (AP) are two factors which contribute to senescence.[7] Both MA, and AP contribute to age-related declines in fitness.[8] The accumulation of random, germline age-related mutated alleles is known as mutation accumulation. Note that somatic mutations are not heritable, they are only a source of developmental variation. Studies done on Drosophila melanogaster have shown that mutation accumulation drives the combination of alleles which have "age-specific additive effects" that cause a decline in stress response and ultimately an age-related decline in fitness.[7] The number of germ cell divisions per generation is variable among lineages, and relates to genome size; for humans; 401 germ cell divisions occur per generation in males and 31 in females.[9]
Mutation accumulation
Germ line
The first modern theory of mammal ageing was formulated by Peter Medawar in 1952. This theory formed in the previous decade with J. B. S. Haldane and his selection shadow concept. The development of human civilization has shifted the selective shadow as the conditions that humans now live in include improved quality of victuals, living conditions, and healthcare. This improved healthcare includes modern medicine such as antibiotics and new medical technology.[10] A few studies in Drosophila have shown that the age of expression of novel deleterious mutations, defines the effects they contribute on mortality. Overall, however; although their frequency increases, their effects and variation decreases with age.
There is no theory that explains how these deleterious mutations affect fitness on different ages and the evolution of senescence.[11][12] Their idea was that ageing was a matter of neglect, as nature is a highly competitive place. Almost all animals die in the wild from predators, disease, or accidents, which lowers the average age of death. Therefore, there is not much reason why the body should remain fit for the long haul because selection pressure is low for traits that would maintain viability past the time when most animals would have died anyway. Metabolic diseases come along due to the low demand for physical activity in modern civilization compared to times where humans had to forage in the wild for survival.[10] With the selective shadow now shifted, humans must deal with these new selective pressures.
Senescence is considered a by-product of physiology because our cell metabolism creates products that are toxic, we get mutations when we age, and we don't have enough stem cells that regenerate. Why did selection not find and favor mutations in ways that allow us, for example, to regenerate our cells, or to not produce toxic metabolism? Why did menopause evolve? Because selection is more efficient on traits that appear early in life. Mutations that have an effect early in life will increase fitness much more than mutations that manifest late. Most people have already reproduced before any disease manifest; this means that parents will pass their alleles to their offsprings before they show any fitness problems, and it is therefore "too late" for selection.
The two theories; non-adaptive, and adaptive, are used to explain the evolution of senescence, which is the decline in reproduction with age.[8] The non-adaptive theory assumes that the evolutionary deterioration of human age occurs as a result of accumulation of deleterious mutations in the germline.[8] These deleterious mutations start expressing themselves late in life, by the time we are weak/wobbly and have already reproduced, this means that Natural selection cannot act on them because reproduction has ended. Studies done on Drosophila melanogaster have shown an inverse relationship between the mean optimal age at maturity and mutation rates per gene.[13] Mutation accumulation affects the allocation of energy, and time that are directed towards growth and reproduction over the lifetime of an organism, especially the period of reproductive lifespan due to the fact that mutation accumulation accelerates senescence, this means that organisms must reach the optimum age of maturity at a younger age as their reproductive lifespan is shortened with accumulated mutation.[13]
Mutations happen, and they are completely random with respect to a need in the environment and fitness. Mutations can either be beneficial in which they increase an organism's fitness, neutral in which they do not affect an organism's fitness or deleterious where they negatively affect an organism's fitness. Previously done experiments have shown that most mutation accumulations are deleterious, and just a few are beneficial. Mutations of genes that interact with one another during the developmental process create biological and, thus, phenotypical diversities. Mutations is genetic information that are expressed among organisms via gene expression, which is the translation of genetic information into a phenotypic character.[14] Evolution is the change in a heritable trait in a population across generations since mutations generate variations in the heritable traits; they are considered the raw material for evolution. Therefore, beneficial mutation accumulations during the developmental processes could generate more phenotypic variations, which increases their gene frequency and affect the capacity of phenotypic evolution.[15]
Somatic cells
The somatic mutation theory of ageing states that accumulation of mutations in somatic cells is the primary cause of aging. A comparison of somatic mutation rate across several mammal species found that the total number of accumulated mutations at the end of lifespan was roughly equal across a broad range of lifespans.[16] The authors state that this strong relationship between somatic mutation rate and lifespan across different mammalian species suggests that evolution may constrain somatic mutation rates, perhaps by selection acting on different DNA repair pathways.
Antagonistic pleiotropy
Medawar's theory was critiqued and later further developed by George C. Williams in 1957. Williams noted that senescence may be causing many deaths even if animals are not 'dying of old age.'[1] He began his hypothesis with the idea that ageing can cause earlier senescence due to the competitive nature of life. Even a small amount of ageing can be fatal; hence natural selection does indeed care and ageing is not cost-free.[17]
Williams eventually proposed his own hypothesis called antagonistic pleiotropy. Pleiotropy, alone, means one mutation that cause multiple effects on phenotype.[18] Antagonistic pleiotropy on the other hand deals with one gene that creates two traits with one being beneficial and the other detrimental. In essence, this refers to genes that offer benefits early in life, but later accumulate a cost.[1] In other words, antagonistic pleiotropy is when the resultant relationship between two traits is negative. It's when one phenotypic trait positively affects current reproduction at the expense of later accelerated senescence, growth, and maintenance. Antagonistic pleiotropy is permanent unless a mutation that modifies the effects of the primary locus occurs.[13]
Although antagonistic pleiotropy is a prevailing theory today, this is largely by default, and has not been well verified. Research has shown that this is not true for all genes and may be thought of as partial validation of the theory, but it cuts the core premise: that genetic trade-offs are the root cause of ageing.
In breeding experiments, Michael R. Rose selected fruit flies for long lifespan. Based on antagonistic pleiotropy, Rose expected that this would surely reduce their fertility. His team found that they were able to breed flies that lived more than twice as long as the flies they started with, but to their surprise, the long-lived, inbred flies actually laid more eggs than the short-lived flies. This was another setback for pleiotropy theory, though Rose maintains it may be an experimental artifact.[19]
Disposable soma theory
A third mainstream theory, proposed in 1977 by Thomas Kirkwood, presumes that the body must budget the resources available to it. The body uses resources derived from the environment for metabolism, for reproduction, and for repair and maintenance, and the body must compromise when there is a finite supply of resources. The theory states that this compromise causes the body to reallocate energy to the repair function that causes the body to gradually deteriorate with age.[2]
A caveat to this theory suggests that this reallocation of energy is based on time instead of limiting resources. This concept focuses on the evolutionary pressure to reproduce in a set, optimal time period that is dictated by age and ecological niche. The way that this is successful is through the allocation of time and energy in damage repair at the cellular level resulting in an accumulation of damage and a decreased lifespan relative to organisms with longer gestation. This concept stems from a comparative analysis of genomic stability in mammalian cells.[20][21]
One opposing argument is based on the effect of caloric restriction, which lengthens life.[22][23][24] However, dietary restriction has not been shown to increase lifetime reproductive success (fitness), because when food availability is lower, reproductive output is also lower. Moreover, calories are not the only resource of possibly limited supply to an organism that could have an effect on multiple dimensions of fitness.
DNA damage/error theory
Just like DNA mutation and expression have phenotypic effects on organisms, DNA damage and mutation accumulation also have phenotypic consequences in older humans. Damage to macromolecules such as DNA, RNA, and proteins along with the deterioration of tissues and organs are the basis of aging. Species-specific rates of aging are due to deleterious changes which manifest after the reproductive phase. "Mitochondrial DNA (mtDNA) regulates cellular metabolism, apoptosis and oxidative stress control".[25] Damage to mtDNA is therefore another contributing factor to phenotypes related to aging. Neurodegeneration and cancer are two factors that manifest with DNA damage; therefore, we need to understand the change in the association between DNA damage and DNA repair as we age in order to be aware of age-related diseases and develop lifestyles that could possibly promote a healthy life span.[26]
The DNA damage theory of aging postulates that DNA damage is ubiquitous in the biological world and is the primary cause of ageing.[27] The theory is based on the idea that ageing occurs over time due to the damage of the DNA. As an example, studies of mammalian brain and muscle have shown that DNA repair capability is relatively high during early development when cells are dividing mitotically, but declines substantially as cells enter the post-mitotic state.[28][29][30]
The effect of reducing expression of DNA repair capability is increased accumulation of DNA damage. This impairs gene transcription and causes the progressive loss of cellular and tissue functions that define aging. As a response to DNA damage, one of the responses triggered by oxidative stress is the activation of the p53.[31] The p53 protein binds to DNA, then stimulates the production of a p21, which is also known as cyclin-dependent kinase inhibitor 1. This ensures that the cell cannot enter the next stage of cell division unless the DNA damage is repaired. However, the p21 cells can trigger apoptosis. Apoptosis or programmed cell death is associated with gradual degradation of the immune system, skeletal muscle, and aging-associated malfunction.[32]

Telomere theory of ageing
Telomeres are recurring nucleotide sequences that protect the ends of our chromosome; they are sensitive to oxidative stress and degrade during chromosomal replication. Telomerase is a ribonucleotide protein that helps repair and replace degraded telomeres. However, telomerase fails us as we age; it becomes less able to repair telomeres, and our whole body starts falling apart. This means that our cells can no longer divide or divide with errors, and some believe that this contributes to the process of aging. New research has also shown that there is an association between telomere shortening and mitochondrial dysfunction.[33] Nevertheless, over-expression of telomerase increases the chances of cancer. If telomeres stay in repair, there is a greater chance of longevity, but there is also more cell division and a greater chance of mutation, which could result in cancer. Therefore, a long-lived cell is just a time bomb. Enhancing telomerase activity is, therefore, not a solution; it only allows the cells to live longer. Naked mole rats have high telomerase activity, they live long, and were thought by some to never get cancer; and therefore possibly be an exception to this hypothesis.[34] Naked mole rats do get cancer, however.[35][36]
Programmed maintenance theories
Theories, such as Weismann's "programmed death" theory, suggest that deterioration and death due to ageing are a purposeful result of an organism's evolved design, and are referred to as theories of programmed ageing or adaptive ageing.
The programmed maintenance theory based on evolvability[37] suggests that the repair mechanisms are controlled by a common control mechanism capable of sensing conditions, such as caloric restriction, and may be responsible for lifespan in particular species. In this theory, the survival techniques are based on control mechanisms instead of individual maintenance mechanism, which you see in the non-programmed theory of mammal ageing.
A non-programmed theory of mammal ageing[38] states that different species possess different capabilities for maintenance and repair. Longer-lived species possess many mechanisms for offsetting damage due to causes such as oxidation, telomere shortening, and other deteriorative processes. Shorter-lived species, having earlier ages of sexual maturity, have less need for longevity and thus did not evolve or retain the more-effective repair mechanisms. Damage therefore accumulates more rapidly, resulting in earlier manifestations and shorter lifespan. Since there are a wide variety of ageing manifestations that appear to have very different causes, it is likely that there are many different maintenance and repair functions.
Selective shadow
Selective shadowing is one of the evolutionary theories of aging based on the presumption that selection of an individual generally decreases once they essentially pass the sexual mature phase. As a result, this forms a shadow without the account of sexual fitness, which is no longer considered as an individual ages. This supports the idea that the force of natural selection declines as a function of age, which was first introduced by Peter B. Medewar and J.B.S Haldane.
"The key conceptual insight that allowed Medawar, Williams, and others, to develop the evolutionary theory of aging is based on the notion that the force of natural selection, a measure of how effectively selection acts on survival rate or fecundity as a function of age, declines with progressive age."[39]
Medewar developed a model that highlights this, showing the decrease in the survival rate of a population as an individual ages, however the reproduction rate stays constant. The reproduction probability typically peaks during sexual maturity and decreases as an individual ages, while the rest of the population decreases with age as they enter the selection shadow. The model also supports Medewars' theory that due to dangerous and unpredicted conditions in the environment such as diseases, climate changes and predators, many individuals die not too long after sexual maturation. Consequently, the probability of an individual surviving and suffering from age related effects is relatively low.
In the same way, many beneficial mutations are selected against if they have a positive effect on an individual later on in life. For instance if a beneficial or deleterious mutation occurs only after an individual's reproductive phase, then it will not affect fitness, which therefore can not be selected against. Subsequently, these later mutations and effects are considered to be in the "shadow region" of selection."[40]
Natural selection
Group selection
Group selection is based on the idea that all members of a given group will either succeed or fail together depending on the circumstance. With this mechanism, genetic drift occurs collectively to all in the group and sets them apart from other groups of its own species. This is different than individual selection, as it focuses on the group rather than the individual.[41]
Often also postreproductive individuals make intergenerational transfers: bottlenose dolphins and pilot whales guard their grandchildren; there is cooperative breeding in some mammals, many insects and about 200 species of birds; sex differences in the survival of anthropoid primates tend to correlate with the care to offspring; or an Efe infant is often attended by more than 10 people. Lee developed a formal theory integrating selection due to transfers (at all ages) with selection due to fertility.[42]
Evolvability
Evolvability is the concept that a species should profit from faster genetic adaptation to its present environment. In the following examples, this is used to argue that eliminating old individuals might benefit the species overall.
Skulachev (1997)[43] has suggested that programmed ageing assists the evolution process by providing a gradually increasing challenge or obstacle to survival and reproduction, and therefore enhancing the selection of beneficial characteristics.
Goldsmith (2008)[44] proposed that though increasing the generation rate and evolution rate is beneficial for a species, it is also important to limit lifespan so older individuals will not dominate the gene pool.
Yang (2013)'s model[6] is also based on the idea that ageing accelerates the accumulation of novel adaptive genes in local populations. However, Yang changed the terminology of "evolvability" into "genetic creativity" throughout his paper to facilitate the understanding of how ageing can have a shorter-term benefit than the word "evolvability" would imply.
Lenart and Vašku (2016) [45] have also invoked evolvability as the main mechanism driving evolution of ageing. However, they proposed that even though the actual rate of aging can be an adaptation the aging itself is inevitable. In other words, evolution can change the speed of aging but some ageing no matter how slow will always occur.
Mortality
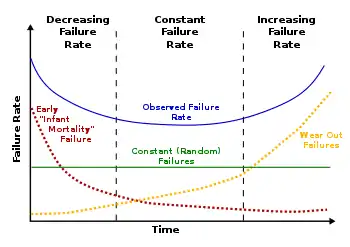
Mortality is the number of deaths, in a particular group, over a specific time period.[46] There are two types of mortality: intrinsic and extrinsic mortality. Intrinsic mortality is defined as mortality due to ageing, the physiological decline due to innate processes, whereas extrinsic mortality is the result of environmental factors such as for example predation, starvation, accidents and others. Flying animals such as bats, for example, have fewer predators, and therefore have a low extrinsic mortality. Birds are warm-blooded and similar in size to many small mammals, yet often live 5–10 times as long. They face less predation than ground-dwelling mammals, and thus have lower extrinsic mortality.
When examining the body-size vs. lifespan relationship, one also observes that predatory mammals tend to live longer than prey mammals in a controlled environment, such as a zoo or nature reserve. The explanation for the long lifespans of primates (such as humans, monkeys, and apes) relative to body size is that due to their intelligence, manage to achieve lower extrinsic mortality.
Potential immortality of the germ line
Individual organisms are ordinarily mortal; they age and die, while the germlines which connect successive generations are potentially immortal. The basis for this difference is a fundamental problem in biology. The Russian biologist and historian Zhores A. Medvedev[47] considered that the accuracy of genome replicative and other synthetic systems alone cannot explain the immortality of germ lines. Rather Medvedev thought that known features of the biochemistry and genetics of sexual reproduction indicate the presence of unique information maintenance and restoration processes at the different stages of gametogenesis. In particular, Medvedev considered that the most important opportunities for information maintenance of germ cells are created by recombination during meiosis and DNA repair; he saw these as processes within the germ cells that were capable of restoring the integrity of DNA and chromosomes from the types of damage that cause irreversible ageing in somatic cells.
Diseases
Progeroid syndromes
Progeroid syndromes are genetic diseases that are linked to premature aging. Progeroid syndromes are characterized by having features that resemble those of physiological aging such as hair loss and cardiovascular disease.[48]
Progeria
Progeria is a single-gene genetic disease that cause acceleration of many or most symptoms of ageing during childhood. It affects about 1 in 4-8 million births.[49] Those who have this disease are known for failure to thrive and have a series of symptoms that cause abnormalities in the joints, hair, skin, eyes, and face.[50] Most who have the disease only live to about age 13.[51] Although the term progeria applies strictly speaking to all diseases characterized by premature aging symptoms, and is often used as such, it is often applied specifically in reference to Hutchinson–Gilford progeria syndrome (HGPS). Children diagnosed with HGPS develop prominent facial features such as a small face, thin lips, small chin, and protruding ears. Although progeria can cause physical abnormalities on a child, it does not impact their motor skills or intellectual advancement.[52] Those who have HGPS are prone to suffer from neurological and cardiovascular disorders.[53] HGPS is caused by a point mutation in the gene that encodes lamin A protein. Lamin A promotes genetic stability by maintaining levels of proteins that have key roles in non-homologous end joining and homologous recombination.[54] Mouse cells deficient for maturation of prelamin A show increased DNA damage and chromosome aberrations and have increased sensitivity to DNA damaging agents.[55] In HGPS, the inability to adequately repair DNA damages due to defective A-type lamin may cause aspects of laminopathy-based premature aging.[55]
Werner Syndrome
Werner syndrome, also known as "adult progeria", is another single-gene genetic disease. it is caused by a mutation in the wrn gene.[53] It affects about 1 in 200,000 people in the United States.[56] This syndrome starts to affect individuals during the teenage years, preventing teens from growing at puberty. There are four common traits of Werner's syndrome: cataracts in both eyes, changes in skin similar to scleroderma, short stature, and early graying and loss of hair.[53] Once the individual reaches the twenties, there is generally a change in hair color, skin, and voice. The average life expectancy of someone with this disease is around 46 years.[57] This condition can also affect the weight distribution between the arms, legs, and torso.[58] Those who have Werner syndrome are at an increased risk for cataracts, type 2 diabetes, different types of cancers, and atherosclerosis.[56] The finding that WRN protein interacts with DNA-PKcs and the Ku protein complex, combined with evidence that WRN deficient cells produce extensive deletions at sites of joining of non-homologous DNA ends, suggests a role for WRN protein in the DNA repair process of non-homologous end joining.[59] WRN protein also appears to play a role in resolving recombination intermediate structures during homologous recombinational repair of DNA double-strand breaks.[59]
Other progeroid syndromes
Bloom syndrome is a rare autosomal recessive disorder that is characterized by short stature, chromosomal instability, predisposition to cancer, and sun-sensitive skin.[60] Those with Bloom syndrome can also have learning disabilities and have an increased risk of developing chronic obstructive pulmonary disease (COPD) and disease.[61]
Cockayne syndrome is a homozygous or heterozygous mutation that results in short stature, abnormalities in head size, and slow growth and development.[62]
Rothmund–Thomson syndrome is a rare autosomal recessive disorder that affects the skin. It is characterized by the sparse hair, juvenile cataracts, skeletal abnormalities, and stunted growth.[63]
Biogerontology
Theories of ageing affect efforts to understand and find treatments for age-related conditions:
- Those who believe in the idea that ageing is an unavoidable side effect of some necessary function (antagonistic pleiotropy or disposable soma theories) logically tend to believe that attempts to delay ageing would result in unacceptable side effects to the necessary functions. Altering ageing is therefore "impossible",[1] and study of ageing mechanisms is of only academic interest.
- Those believing in default theories of multiple maintenance mechanisms tend to believe that ways might be found to enhance the operation of some of those mechanisms. Perhaps they can be assisted by antioxidants or other agents.
- Those who believe in programmed ageing suppose that ways might be found to interfere with the operation of the part of the ageing mechanism that appears to be common to multiple symptoms, essentially "slowing down the clock" and delaying multiple manifestations. Such effect might be obtained by fooling a sense function. One such effort is an attempt to find a "mimetic" that would "mime" the anti-ageing effect of calorie restriction without having to actually radically restrict diet.[64]
See also
References
- Williams GC (December 1957). "Pleiotropy, Natural Selection, and the Evolution of Senescence". Evolution. 11 (4): 398–411. doi:10.1111/j.1558-5646.1957.tb02911.x. JSTOR 2406060. S2CID 84556488.
- Kirkwood TB (November 1977). "Evolution of ageing". Nature. 270 (5635): 301–304. Bibcode:1977Natur.270..301K. doi:10.1038/270301a0. PMID 593350. S2CID 492012.
- Medawar PB (1952). An Unsolved Problem of Biology. Published for the college by H. K. Lewis, London
- Johnson AA, Shokhirev MN, Shoshitaishvili B (November 2019). "Revamping the evolutionary theories of aging". Ageing Research Reviews. 55: 100947. doi:10.1016/j.arr.2019.100947. PMID 31449890.
- Weismann A (1889). Essays upon heredity and kindred biological problems. Oxford: Clarendon Press. Work that describes Weismann's theory about making room for the young.
- Yang JN (2013). "Viscous populations evolve altruistic programmed ageing in ability conflict in a changing environment". Evolutionary Ecology Research. 15: 527–543.
- Everman ER, Morgan TJ (February 2018). "Antagonistic pleiotropy and mutation accumulation contribute to age-related decline in stress response". Evolution; International Journal of Organic Evolution. 72 (2): 303–317. doi:10.1111/evo.13408. PMID 29214647.
- Charlesworth B (May 2001). "Patterns of age-specific means and genetic variances of mortality rates predicted by the mutation-accumulation theory of ageing". Journal of Theoretical Biology. 210 (1): 47–65. Bibcode:2001JThBi.210...47C. doi:10.1006/jtbi.2001.2296. PMID 11343430.
- Drost JB, Lee WR (1995). "Biological basis of germline mutation: comparisons of spontaneous germline mutation rates among drosophila, mouse, and human". Environmental and Molecular Mutagenesis. 25 (Suppl 26): 48–64. doi:10.1002/em.2850250609. PMID 7789362. S2CID 41023021.
- Flatt T, Partridge L (August 2018). "Horizons in the evolution of aging". BMC Biology. 16 (1): 93. doi:10.1186/s12915-018-0562-z. PMC 6100731. PMID 30124168.
- Moorad JA, Promislow DE (August 2008). "A theory of age-dependent mutation and senescence". Genetics. 179 (4): 2061–2073. doi:10.1534/genetics.108.088526. PMC 2516080. PMID 18660535.
- Kraemer SA, Böndel KB, Ness RW, Keightley PD, Colegrave N (December 2017). "Fitness change in relation to mutation number in spontaneous mutation accumulation lines of Chlamydomonas reinhardtii". Evolution; International Journal of Organic Evolution. 71 (12): 2918–2929. doi:10.1111/evo.13360. PMC 5765464. PMID 28884790.
- Dańko MJ, Kozłowski J, Vaupel JW, Baudisch A (2012-04-06). "Mutation accumulation may be a minor force in shaping life history traits". PLOS ONE. 7 (4): e34146. Bibcode:2012PLoSO...734146D. doi:10.1371/journal.pone.0034146. PMC 3320907. PMID 22493680.
- Rifkin SA, Houle D, Kim J, White KP (November 2005). "A mutation accumulation assay reveals a broad capacity for rapid evolution of gene expression". Nature. 438 (7065): 220–223. Bibcode:2005Natur.438..220R. doi:10.1038/nature04114. PMID 16281035. S2CID 4413696.
- Nei M (July 2007). "The new mutation theory of phenotypic evolution". Proceedings of the National Academy of Sciences of the United States of America. 104 (30): 12235–12242. Bibcode:2007PNAS..10412235N. doi:10.1073/pnas.0703349104. PMC 1941456. PMID 17640887.
- Cagan, Alex; Baez-Ortega, Adrian; Brzozowska, Natalia; Abascal, Federico; Coorens, Tim H. H.; Sanders, Mathijs A.; Lawson, Andrew R. J.; Harvey, Luke M. R.; Bhosle, Shriram; Jones, David; Alcantara, Raul E. (April 2022). "Somatic mutation rates scale with lifespan across mammals". Nature. 604 (7906): 517–524. Bibcode:2022Natur.604..517C. doi:10.1038/s41586-022-04618-z. ISSN 1476-4687. PMC 9021023. PMID 35418684.
- Carter AJ, Nguyen AQ (December 2011). "Antagonistic pleiotropy as a widespread mechanism for the maintenance of polymorphic disease alleles". BMC Medical Genetics. 12: 160. doi:10.1186/1471-2350-12-160. PMC 3254080. PMID 22151998.
- Curtsinger JW (2001). "Senescence: Genetic Theories". International Encyclopedia of the Social & Behavioral Sciences. pp. 13897–902. doi:10.1016/B0-08-043076-7/03374-X. ISBN 978-0-08-043076-8.
- Leroi AM, Chippindale AK, Rose MR (August 1994). "Long-term laboratory evolution of a genetic life-history tradeoff in Drosophila melanogaster. 1. The role of genotype-by-environment interaction". Evolution; International Journal of Organic Evolution. 48 (4): 1244–1257. doi:10.1111/j.1558-5646.1994.tb05309.x. PMID 28564485. S2CID 22492109.
- Lorenzini A, Stamato T, Sell C (November 2011). "The disposable soma theory revisited: time as a resource in the theories of aging". Cell Cycle. 10 (22): 3853–3856. doi:10.4161/cc.10.22.18302. PMID 22071624.
- van den Heuvel J, English S, Uller T (2016-01-11). Criscuolo F (ed.). "Disposable Soma Theory and the Evolution of Maternal Effects on Ageing". PLOS ONE. 11 (1): e0145544. Bibcode:2016PLoSO..1145544V. doi:10.1371/journal.pone.0145544. PMC 4709080. PMID 26752635.
- Weindruch R, Walford IL (1986). The Retardation of Aging and Disease by Dietary Restriction. Springfield, IL: Thomas.
- Weindruch R (1996). "The retardation of aging by caloric restriction: studies in rodents and primates". Toxicologic Pathology. 24 (6): 742–745. doi:10.1177/019262339602400618. PMID 8994305. S2CID 13212021.
- Masoro EJ (September 2005). "Overview of caloric restriction and ageing". Mechanisms of Ageing and Development. 126 (9): 913–922. doi:10.1016/j.mad.2005.03.012. PMID 15885745. S2CID 8451228. Overview of caloric restriction and aging.
- Atig RK, Hsouna S, Beraud-Colomb E, Abdelhak S (2009). "[Mitochondrial DNA: properties and applications]". Archives de l'Institut Pasteur de Tunis. 86 (1–4): 3–14. PMID 20707216.
- Maynard S, Fang EF, Scheibye-Knudsen M, Croteau DL, Bohr VA (September 2015). "DNA Damage, DNA Repair, Aging, and Neurodegeneration". Cold Spring Harbor Perspectives in Medicine. 5 (10): a025130. doi:10.1101/cshperspect.a025130. PMC 4588127. PMID 26385091.
- Gensler HL, Bernstein H (September 1981). "DNA damage as the primary cause of aging". The Quarterly Review of Biology. 56 (3): 279–303. doi:10.1086/412317. PMID 7031747. S2CID 20822805.
- Gensler HL (1981). "Low level of U.V.-induced unscheduled DNA synthesis in postmitotic brain cells of hamsters: possible relevance to aging". Experimental Gerontology. 16 (2): 199–207. doi:10.1016/0531-5565(81)90046-2. PMID 7286098. S2CID 6261990.
- Karran P, Moscona A, Strauss B (July 1977). "Developmental decline in DNA repair in neural retina cells of chick embryos. Persistent deficiency of repair competence in a cell line derived from late embryos". The Journal of Cell Biology. 74 (1): 274–286. doi:10.1083/jcb.74.1.274. PMC 2109876. PMID 559680.
- Lampidis TJ, Schaiberger GE (December 1975). "Age-related loss of DNA repair synthesis in isolated rat myocardial cells". Experimental Cell Research. 96 (2): 412–416. doi:10.1016/0014-4827(75)90276-1. PMID 1193184.
- Rodier F, Campisi J, Bhaumik D (2007-12-15). "Two faces of p53: aging and tumor suppression". Nucleic Acids Research. 35 (22): 7475–7484. doi:10.1093/nar/gkm744. PMC 2190721. PMID 17942417.
- Tower J (September 2015). "Programmed cell death in aging". Ageing Research Reviews. 23 (Pt A): 90–100. doi:10.1016/j.arr.2015.04.002. PMC 4480161. PMID 25862945.
- Sahin E, Colla S, Liesa M, Moslehi J, Müller FL, Guo M, et al. (February 2011). "Telomere dysfunction induces metabolic and mitochondrial compromise". Nature. 470 (7334): 359–365. Bibcode:2011Natur.470..359S. doi:10.1038/nature09787. PMC 3741661. PMID 21307849.
- Petruseva IO, Evdokimov AN, Lavrik OI (2017). "Genome Stability Maintenance in Naked Mole-Rat". Acta Naturae. 9 (4): 31–41. doi:10.32607/20758251-2017-9-4-31-41. PMC 5762826. PMID 29340215.
- Delaney, M. A.; Ward, J. M.; Walsh, T. F.; Chinnadurai, S. K.; Kerns, K.; Kinsel, M. J.; Treuting, P. M. (2016). "Initial Case Reports of Cancer in Naked Mole-rats (Heterocephalus glaber)". Veterinary Pathology. 53 (3): 691–696. doi:10.1177/0300985816630796. ISSN 0300-9858. PMID 26846576. S2CID 3746533.
- Taylor, Kyle R.; Milone, Nicholas A.; Rodriguez, Carlos E. (2016). "Four Cases of Spontaneous Neoplasia in the Naked Mole-Rat (Heterocephalus glaber), A Putative Cancer-Resistant Species". The Journals of Gerontology Series A: Biological Sciences and Medical Sciences. 72 (1): 38–43. doi:10.1093/gerona/glw047. ISSN 1079-5006. PMID 27129918.
- Goldsmith T (2009). "Mammal aging: active and passive mechanisms". Journal of Bioscience Hypotheses. 2 (2): 59–64. doi:10.1016/j.bihy.2008.12.002. Article compares programmed and non-programmed maintenance theories of ageing in light of empirical evidence.
- Holliday R (May 2006). "Aging is no longer an unsolved problem in biology". Annals of the New York Academy of Sciences. 1067 (1): 1–9. Bibcode:2006NYASA1067....1H. doi:10.1196/annals.1354.002. PMID 16803964. S2CID 9390016.
- Hamilton WD (September 1966). "The moulding of senescence by natural selection". Journal of Theoretical Biology. 12 (1): 12–45. Bibcode:1966JThBi..12...12H. doi:10.1016/0022-5193(66)90184-6. PMID 6015424.
- Flatt T, Schmidt PS (October 2009). "Integrating evolutionary and molecular genetics of aging". Biochimica et Biophysica Acta (BBA) - General Subjects. 1790 (10): 951–962. doi:10.1016/j.bbagen.2009.07.010. PMC 2972575. PMID 19619612.
- Mitteldorf J (2006). "Chaotic population dynamics and the evolution of ageing: proposing a demographic theory of senescence". Evolutionary Ecology Research. 8: 561–74. On population dynamics as a mechanism for the evolution of ageing.
- Lee RD (August 2003). "Rethinking the evolutionary theory of aging: transfers, not births, shape senescence in social species". Proceedings of the National Academy of Sciences of the United States of America. 100 (16): 9637–9642. Bibcode:2003PNAS..100.9637L. doi:10.1073/pnas.1530303100. PMC 170970. PMID 12878733.
- Skulachev VP (November 1997). "Aging is a specific biological function rather than the result of a disorder in complex living systems: biochemical evidence in support of Weismann's hypothesis". Biochemistry. Biokhimiia. 62 (11): 1191–1195. PMID 9467841.
- Goldsmith TC (June 2008). "Aging, evolvability, and the individual benefit requirement; medical implications of aging theory controversies". Journal of Theoretical Biology. 252 (4): 764–768. Bibcode:2008JThBi.252..764G. doi:10.1016/j.jtbi.2008.02.035. PMID 18396295.
- Lenart P, Bienertová-Vašků J (August 2017). "Keeping up with the Red Queen: the pace of aging as an adaptation". Biogerontology. 18 (4): 693–709. doi:10.1007/s10522-016-9674-4. PMID 28013399. S2CID 11048849.
- "NCI Dictionary of Cancer Terms". National Cancer Institute. 2011-02-02. Retrieved 2020-04-11.
- Medvedev ZA (December 1981). "On the immortality of the germ line: genetic and biochemical mechanism. A review". Mechanisms of Ageing and Development. 17 (4): 331–359. doi:10.1016/0047-6374(81)90052-X. PMID 6173551. S2CID 35719466.
- Carrero D, Soria-Valles C, López-Otín C (July 2016). "Hallmarks of progeroid syndromes: lessons from mice and reprogrammed cells". Disease Models & Mechanisms. 9 (7): 719–735. doi:10.1242/dmm.024711. PMC 4958309. PMID 27482812.
- "Progeria". WebMD. Retrieved 2020-04-11.
- "Hutchinson-Gilford progeria syndrome". Genetics Home Reference. Retrieved 2019-03-27.
- King RC, Mulligan PK, Stansfield WD (2013). A dictionary of genetics (8th ed.). New York: Oxford University Press. ISBN 978-0-19-937686-5. OCLC 871046520.
- "Hutchinson-Gilford progeria syndrome". Genetics Home Reference. Retrieved 2020-04-11.
- McDonald RB (2019). Biology of aging (Second ed.). Boca Raton. ISBN 978-0-8153-4567-1. OCLC 1056201427.
{{cite book}}: CS1 maint: location missing publisher (link) - Redwood AB, Perkins SM, Vanderwaal RP, Feng Z, Biehl KJ, Gonzalez-Suarez I, Morgado-Palacin L, Shi W, Sage J, Roti-Roti JL, Stewart CL, Zhang J, Gonzalo S (27 October 2014). "A dual role for A-type lamins in DNA double-strand break repair". Cell Cycle. 10 (15): 2549–2560. doi:10.4161/cc.10.15.16531. PMC 3180193. PMID 21701264
- Liu B, Wang J, Chan KM, Tjia WM, Deng W, Guan X, Huang J-d, Li KM, Chau PY, Chen DJ, Pei D, Pendas AM, Cadiñanos J, López-Otín C, Tse HF, Hutchison C, Chen J, Cao Y, Cheah KSE, Tryggvason K, Zhou Z (26 June 2005). "Genomic instability in laminopathy-based premature aging". Nature Medicine. 11 (7): 780–785. doi:10.1038/nm1266. PMID 15980864. S2CID 11798376
- "Werner syndrome". Genetics Home Reference. Retrieved 2020-04-11.
- Yamamoto K, Imakiire A, Miyagawa N, Kasahara T (December 2003). "A report of two cases of Werner's syndrome and review of the literature". Journal of Orthopaedic Surgery. 11 (2): 224–233. doi:10.1177/230949900301100222. PMID 14676353.
- Navarro CL, Cau P, Lévy N (October 2006). "Molecular bases of progeroid syndromes". Human Molecular Genetics. 15 Spec No 2 (suppl_2): R151–R161. doi:10.1093/hmg/ddl214. PMID 16987878.
- Thompson LH, Schild D. Recombinational DNA repair and human disease. Mutat Res. 2002 Nov 30;509(1-2):49-78. doi: 10.1016/s0027-5107(02)00224-5. PMID 12427531
- "OMIM Entry - # 210900 - BLOOM SYNDROME; BLM". omim.org. Retrieved 2020-04-11.
- "Bloom syndrome". Genetics Home Reference. Retrieved 2020-04-11.
- "OMIM Entry - # 216400 - COCKAYNE SYNDROME A; CSA". omim.org. Retrieved 2020-04-11.
- "Rothmund-Thomson syndrome". Genetics Home Reference. Retrieved 2020-04-11.
- Chen D, Guarente L (February 2007). "SIR2: a potential target for calorie restriction mimetics". Trends in Molecular Medicine. 13 (2): 64–71. doi:10.1016/j.molmed.2006.12.004. PMID 17207661.
Further reading
- Alcock J (2017). "Human Sociobiology and Group Selection Theory". On Human Nature Biology, Psychology, Ethics, Politics, and Religion. pp. 383–396. doi:10.1016/b978-0-12-420190-3.00023-5. ISBN 978-0-12-420190-3.
{{cite book}}:|work=ignored (help) - Jin K (October 2010). "Modern Biological Theories of Aging". Aging and Disease. 1 (2): 72–74. PMC 2995895. PMID 21132086.
- Gavrilova NS, Gavrilov LA, Semyonova VG, Evdokushkina GN (June 2004). "Does exceptional human longevity come with a high cost of infertility? Testing the evolutionary theories of aging". Annals of the New York Academy of Sciences. 1019 (1): 513–517. Bibcode:2004NYASA1019..513G. CiteSeerX 10.1.1.10.7390. doi:10.1196/annals.1297.095. PMID 15247077. S2CID 10335962.
- Gavrilova NS, Gavrilov LA (2005). "Human longevity and reproduction: An evolutionary perspective.". In Voland E, Chasiotis A, Schiefenhoevel W (eds.). Grandmotherhood - The Evolutionary Significance of the Second Half of Female Life. New Brunswick, NJ, USA: Rutgers University Press. pp. 59–80.
- Gavrilova NS, Gavrilov LA (2002). "Evolution of Aging". In Ekerdt DJ (ed.). Encyclopedia of Aging. Vol. 2. New York: Macmillan Reference USA. pp. 458–467.
- Gavrilov LA, Gavrilova NS (February 2002). "Evolutionary theories of aging and longevity". TheScientificWorldJournal. 2: 339–356. doi:10.1100/tsw.2002.96. PMC 6009642. PMID 12806021.
- Gavrilova NS, Gavrilov LA, Evdokushkina GN, Semyonova VG, Gavrilova AL, Evdokushkina NN, et al. (August 1998). "Evolution, mutations, and human longevity: European royal and noble families". Human Biology. 70 (4): 799–804. PMID 9686488.
External links
- Evolutionary Theories of Aging and Longevity
- The Evolutionary Theory of Aging by João Pedro de Magalhães.
- Programmed-Aging.Org Site provides comprehensive information on programmed ageing, the programmed/non-programmed controversy, and underlying evolution controversies.
- How Evolutionary Thinking Affects People's Ideas About Aging Interventions
- AnAge Animal Ageing and Longevity Database Provides maximum observed ages and sexual maturity ages for many animals.
- The Case for Programmed Mammal Aging Describes empirical data, evolutionary rationale, and historical perspective supporting programmed ageing in mammals.
- Life Table for USA 2005 Probability of death as a function of age