Structure factor
In condensed matter physics and crystallography, the static structure factor (or structure factor for short) is a mathematical description of how a material scatters incident radiation. The structure factor is a critical tool in the interpretation of scattering patterns (interference patterns) obtained in X-ray, electron and neutron diffraction experiments.
Confusingly, there are two different mathematical expressions in use, both called 'structure factor'. One is usually written ; it is more generally valid, and relates the observed diffracted intensity per atom to that produced by a single scattering unit. The other is usually written or and is only valid for systems with long-range positional order — crystals. This expression relates the amplitude and phase of the beam diffracted by the planes of the crystal ( are the Miller indices of the planes) to that produced by a single scattering unit at the vertices of the primitive unit cell. is not a special case of ; gives the scattering intensity, but gives the amplitude. It is the modulus squared that gives the scattering intensity. is defined for a perfect crystal, and is used in crystallography, while is most useful for disordered systems. For partially ordered systems such as crystalline polymers there is obviously overlap, and experts will switch from one expression to the other as needed.





The static structure factor is measured without resolving the energy of scattered photons/electrons/neutrons. Energy-resolved measurements yield the dynamic structure factor.
Derivation of S(q)
Consider the scattering of a beam of wavelength by an assembly of particles or atoms stationary at positions . Assume that the scattering is weak, so that the amplitude of the incident beam is constant throughout the sample volume (Born approximation), and absorption, refraction and multiple scattering can be neglected (kinematic diffraction). The direction of any scattered wave is defined by its scattering vector . , where and ( ) are the scattered and incident beam wavevectors, and is the angle between them. For elastic scattering, and , limiting the possible range of (see Ewald sphere). The amplitude and phase of this scattered wave will be the vector sum of the scattered waves from all the atoms [1][2]












For an assembly of atoms, is the atomic form factor of the -th atom. The scattered intensity is obtained by multiplying this function by its complex conjugate


-
(1)

The structure factor is defined as this intensity normalized by [3]

-
(2)

If all the atoms are identical, then Equation (1) becomes and so


-
(3)

Another useful simplification is if the material is isotropic, like a powder or a simple liquid. In that case, the intensity depends on and . In three dimensions, Equation (2) then simplifies to the Debye scattering equation:[1]


-
(4)

An alternative derivation gives good insight, but uses Fourier transforms and convolution. To be general, consider a scalar (real) quantity defined in a volume ; this may correspond, for instance, to a mass or charge distribution or to the refractive index of an inhomogeneous medium. If the scalar function is integrable, we can write its Fourier transform as . In the Born approximation the amplitude of the scattered wave corresponding to the scattering vector is proportional to the Fourier transform .[1] When the system under study is composed of a number of identical constituents (atoms, molecules, colloidal particles, etc.) each of which has a distribution of mass or charge then the total distribution can be considered the convolution of this function with a set of delta functions.





-
(5)

with the particle positions as before. Using the property that the Fourier transform of a convolution product is simply the product of the Fourier transforms of the two factors, we have , so that:

-
(6)

This is clearly the same as Equation (1) with all particles identical, except that here is shown explicitly as a function of .

In general, the particle positions are not fixed and the measurement takes place over a finite exposure time and with a macroscopic sample (much larger than the interparticle distance). The experimentally accessible intensity is thus an averaged one ; we need not specify whether denotes a time or ensemble average. To take this into account we can rewrite Equation (3) as:


-
(7)

Perfect crystals
In a crystal, the constitutive particles are arranged periodically, with translational symmetry forming a lattice. The crystal structure can be described as a Bravais lattice with a group of atoms, called the basis, placed at every lattice point; that is, [crystal structure] = [lattice] [basis]. If the lattice is infinite and completely regular, the system is a perfect crystal. For such a system, only a set of specific values for can give scattering, and the scattering amplitude for all other values is zero. This set of values forms a lattice, called the reciprocal lattice, which is the Fourier transform of the real-space crystal lattice.

In principle the scattering factor can be used to determine the scattering from a perfect crystal; in the simple case when the basis is a single atom at the origin (and again neglecting all thermal motion, so that there is no need for averaging) all the atoms have identical environments. Equation (1) can be written as
- and .


The structure factor is then simply the squared modulus of the Fourier transform of the lattice, and shows the directions in which scattering can have non-zero intensity. At these values of the wave from every lattice point is in phase. The value of the structure factor is the same for all these reciprocal lattice points, and the intensity varies only due to changes in with .
Units
The units of the structure-factor amplitude depend on the incident radiation. For X-ray crystallography they are multiples of the unit of scattering by a single electron (2.82 m); for neutron scattering by atomic nuclei the unit of scattering length of m is commonly used.


The above discussion uses the wave vectors and . However, crystallography often uses wave vectors and . Therefore, when comparing equations from different sources, the factor may appear and disappear, and care to maintain consistent quantities is required to get correct numerical results.





Definition of Fhkl
In crystallography, the basis and lattice are treated separately. For a perfect crystal the lattice gives the reciprocal lattice, which determines the positions (angles) of diffracted beams, and the basis gives the structure factor which determines the amplitude and phase of the diffracted beams:

-
(8)
![{\displaystyle F_{hk\ell }=\sum _{j=1}^{N}f_{j}\mathrm {e} ^{[-2\pi i(hx_{j}+ky_{j}+\ell z_{j})]},}](../I/16ee72ccd9ba16412dd9d2a2c62a6041d740f2fb.svg)
where the sum is over all atoms in the unit cell, are the positional coordinates of the -th atom, and is the scattering factor of the -th atom.[4] The coordinates have the directions and dimensions of the lattice vectors . That is, (0,0,0) is at the lattice point, the origin of position in the unit cell; (1,0,0) is at the next lattice point along and (1/2, 1/2, 1/2) is at the body center of the unit cell. defines a reciprocal lattice point at which corresponds to the real-space plane defined by the Miller indices (see Bragg's law).





is the vector sum of waves from all atoms within the unit cell. An atom at any lattice point has the reference phase angle zero for all since then is always an integer. A wave scattered from an atom at (1/2, 0, 0) will be in phase if is even, out of phase if is odd.



Again an alternative view using convolution can be helpful. Since [crystal structure] = [lattice] [basis], [crystal structure] = [lattice] [basis]; that is, scattering [reciprocal lattice] [structure factor].




Body-centered cubic (BCC)
For the body-centered cubic Bravais lattice (cI), we use the points and which leads us to


![{\displaystyle F_{hk\ell }=\sum _{j}f_{j}e^{-2\pi i(hx_{j}+ky_{j}+\ell z_{j})}=f\left[1+\left(e^{-i\pi }\right)^{h+k+\ell }\right]=f\left[1+(-1)^{h+k+\ell }\right]}](../I/04e041de2449b496dd9c30c6713e3a989ca5a7c8.svg)
and hence

Face-centered cubic (FCC)
The FCC lattice is a Bravais lattice, and its Fourier transform is a body-centered cubic lattice. However to obtain without this shortcut, consider an FCC crystal with one atom at each lattice point as a primitive or simple cubic with a basis of 4 atoms, at the origin and at the three adjacent face centers, , and . Equation (8) becomes




![{\displaystyle F_{hk\ell }=f\sum _{j=1}^{4}\mathrm {e} ^{[-2\pi i(hx_{j}+ky_{j}+\ell z_{j})]}=f\left[1+\mathrm {e} ^{[-i\pi (h+k)]}+\mathrm {e} ^{[-i\pi (k+\ell )]}+\mathrm {e} ^{[-i\pi (h+\ell )]}\right]=f\left[1+(-1)^{h+k}+(-1)^{k+\ell }+(-1)^{h+\ell }\right]}](../I/04c11968ee184e993a75bdbc1eb6ad5c6091b351.svg)
with the result

The most intense diffraction peak from a material that crystallizes in the FCC structure is typically the (111). Films of FCC materials like gold tend to grow in a (111) orientation with a triangular surface symmetry. A zero diffracted intensity for a group of diffracted beams (here, of mixed parity) is called a systematic absence.

Diamond crystal structure
The diamond cubic crystal structure occurs for example diamond (carbon), tin, and most semiconductors. There are 8 atoms in the cubic unit cell. We can consider the structure as a simple cubic with a basis of 8 atoms, at positions

But comparing this to the FCC above, we see that it is simpler to describe the structure as FCC with a basis of two atoms at (0, 0, 0) and (1/4, 1/4, 1/4). For this basis, Equation (8) becomes:
![{\displaystyle F_{hk\ell }({\rm {{basis})=f\sum _{j=1}^{2}\mathrm {e} ^{[-2\pi i(hx_{j}+ky_{j}+\ell z_{j})]}=f\left[1+\mathrm {e} ^{[-i\pi /2(h+k+\ell )]}\right]=f\left[1+(-i)^{h+k+\ell }\right]}}}](../I/fec74575f85a623a6d38001ff0d15abc5f0ab1cb.svg)
And then the structure factor for the diamond cubic structure is the product of this and the structure factor for FCC above, (only including the atomic form factor once)
![{\displaystyle F_{hk\ell }=f\left[1+(-1)^{h+k}+(-1)^{k+\ell }+(-1)^{h+\ell }\right]\times \left[1+(-i)^{h+k+\ell }\right]}](../I/b671c5a02c9f1655e5bd9e2e93552c4b32ca9c9f.svg)
with the result
- If h, k, ℓ are of mixed parity (odd and even values combined) the first (FCC) term is zero, so
- If h, k, ℓ are all even or all odd then the first (FCC) term is 4
- if h+k+ℓ is odd then
- if h+k+ℓ is even and exactly divisible by 4 () then
- if h+k+ℓ is even but not exactly divisible by 4 () the second term is zero and





These points are encapsulated by the following equations:


where is an integer.
Zincblende crystal structure
The zincblende structure is similar to the diamond structure except that it is a compound of two distinct interpenetrating fcc lattices, rather than all the same element. Denoting the two elements in the compound by and , the resulting structure factor is



Cesium chloride
Cesium chloride is a simple cubic crystal lattice with a basis of Cs at (0,0,0) and Cl at (1/2, 1/2, 1/2) (or the other way around, it makes no difference). Equation (8) becomes
![{\displaystyle F_{hk\ell }=\sum _{j=1}^{2}f_{j}\mathrm {e} ^{[-2\pi i(hx_{j}+ky_{j}+\ell z_{j})]}=\left[f_{Cs}+f_{Cl}\mathrm {e} ^{[-i\pi (h+k+\ell )]}\right]=\left[f_{Cs}+f_{Cl}(-1)^{h+k+\ell }\right]}](../I/2c30153f90296fb37f56f345a9d5d3cfb1555d03.svg)
We then arrive at the following result for the structure factor for scattering from a plane :

and for scattered intensity,

Hexagonal close-packed (HCP)
In an HCP crystal such as graphite, the two coordinates include the origin and the next plane up the c axis located at c/2, and hence , which gives us


![{\displaystyle F_{hk\ell }=f\left[1+e^{2\pi i\left({\tfrac {h}{3}}+{\tfrac {2k}{3}}+{\tfrac {\ell }{2}}\right)}\right]}](../I/a4ef61f01b9a8073319184e64f373da4e483abf8.svg)
From this it is convenient to define dummy variable , and from there consider the modulus squared so hence

![{\displaystyle |F|^{2}=f^{2}\left(1+e^{2\pi iX}\right)\left(1+e^{-2\pi iX}\right)=f^{2}\left(2+e^{2\pi iX}+e^{-2\pi iX}\right)=f^{2}\left(2+2\cos[2\pi X]\right)=f^{2}\left(4\cos ^{2}\left[\pi X\right]\right)}](../I/d213a802f67c2f42886cdb86f5c54ac22aa461ca.svg)
This leads us to the following conditions for the structure factor:

Perfect crystals in one and two dimensions
The reciprocal lattice is easily constructed in one dimension: for particles on a line with a period , the reciprocal lattice is an infinite array of points with spacing . In two dimensions, there are only five Bravais lattices. The corresponding reciprocal lattices have the same symmetry as the direct lattice. 2-D lattices are excellent for demonstrating simple diffraction geometry on a flat screen, as below. Equations (1)–(7) for structure factor apply with a scattering vector of limited dimensionality and a crystallographic structure factor can be defined in 2-D as .


![{\displaystyle F_{hk}=\sum _{j=1}^{N}f_{j}\mathrm {e} ^{[-2\pi i(hx_{j}+ky_{j})]}}](../I/5ffdca0e7ed5ef03fd54745d58831056abbde955.svg)
However, recall that real 2-D crystals such as graphene exist in 3-D. The reciprocal lattice of a 2-D hexagonal sheet that exists in 3-D space in the plane is a hexagonal array of lines parallel to the or axis that extend to and intersect any plane of constant in a hexagonal array of points.




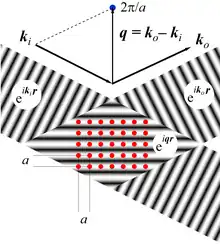


The Figure shows the construction of one vector of a 2-D reciprocal lattice and its relation to a scattering experiment.
A parallel beam, with wave vector is incident on a square lattice of parameter . The scattered wave is detected at a certain angle, which defines the wave vector of the outgoing beam, (under the assumption of elastic scattering, ). One can equally define the scattering vector and construct the harmonic pattern . In the depicted example, the spacing of this pattern coincides to the distance between particle rows: , so that contributions to the scattering from all particles are in phase (constructive interference). Thus, the total signal in direction is strong, and belongs to the reciprocal lattice. It is easily shown that this configuration fulfills Bragg's law.




Imperfect crystals
Technically a perfect crystal must be infinite, so a finite size is an imperfection. Real crystals always exhibit imperfections of their order besides their finite size, and these imperfections can have profound effects on the properties of the material. André Guinier[5] proposed a widely employed distinction between imperfections that preserve the long-range order of the crystal that he called disorder of the first kind and those that destroy it called disorder of the second kind. An example of the first is thermal vibration; an example of the second is some density of dislocations.
The generally applicable structure factor can be used to include the effect of any imperfection. In crystallography, these effects are treated as separate from the structure factor , so separate factors for size or thermal effects are introduced into the expressions for scattered intensity, leaving the perfect crystal structure factor unchanged. Therefore, a detailed description of these factors in crystallographic structure modeling and structure determination by diffraction is not appropriate in this article.
Finite-size effects
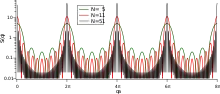
For a finite crystal means that the sums in equations 1-7 are now over a finite . The effect is most easily demonstrated with a 1-D lattice of points. The sum of the phase factors is a geometric series and the structure factor becomes:

![{\displaystyle S(q)={\frac {1}{N}}\left|{\frac {1-\mathrm {e} ^{-iNqa}}{1-\mathrm {e} ^{-iqa}}}\right|^{2}={\frac {1}{N}}\left[{\frac {\sin(Nqa/2)}{\sin(qa/2)}}\right]^{2}.}](../I/a16c0940db45302937cbdcc5096e09394d3b3d53.svg)
This function is shown in the Figure for different values of . When the scattering from every particle is in phase, which is when the scattering is at a reciprocal lattice point , the sum of the amplitudes must be and so the maxima in intensity are . Taking the above expression for and estimating the limit using, for instance, L'Hôpital's rule) shows that as seen in the Figure. At the midpoint (by direct evaluation) and the peak width decreases like . In the large limit, the peaks become infinitely sharp Dirac delta functions, the reciprocal lattice of the perfect 1-D lattice.







In crystallography when is used, is large, and the formal size effect on diffraction is taken as , which is the same as the expression for above near to the reciprocal lattice points, . Using convolution, we can describe the finite real crystal structure as [lattice] [basis] rectangular function, where the rectangular function has a value 1 inside the crystal and 0 outside it. Then [crystal structure] = [lattice] [basis] [rectangular function]; that is, scattering [reciprocal lattice] [structure factor] [ sinc function]. Thus the intensity, which is a delta function of position for the perfect crystal, becomes a function around every point with a maximum , a width , area .
![{\displaystyle \left[{\frac {\sin(Nqa/2)}{(qa/2)}}\right]^{2}}](../I/38d2910fac064c5c2c6c4010de4e47aaa70b21ff.svg)




Disorder of the first kind
This model for disorder in a crystal starts with the structure factor of a perfect crystal. In one-dimension for simplicity and with N planes, we then start with the expression above for a perfect finite lattice, and then this disorder only changes by a multiplicative factor, to give[1]
![{\displaystyle S(q)={\frac {1}{N}}\left[{\frac {\sin(Nqa/2)}{\sin(qa/2)}}\right]^{2}\exp \left(-q^{2}\langle \delta x^{2}\rangle \right)}](../I/06fe65c55c7a9ead569dfeb164efdec6e5c0e150.svg)
where the disorder is measured by the mean-square displacement of the positions from their positions in a perfect one-dimensional lattice: , i.e., , where is a small (much less than ) random displacement. For disorder of the first kind, each random displacement is independent of the others, and with respect to a perfect lattice. Thus the displacements do not destroy the translational order of the crystal. This has the consequence that for infinite crystals () the structure factor still has delta-function Bragg peaks – the peak width still goes to zero as , with this kind of disorder. However, it does reduce the amplitude of the peaks, and due to the factor of in the exponential factor, it reduces peaks at large much more than peaks at small .







The structure is simply reduced by a and disorder dependent term because all disorder of the first-kind does is smear out the scattering planes, effectively reducing the form factor.
In three dimensions the effect is the same, the structure is again reduced by a multiplicative factor, and this factor is often called the Debye–Waller factor. Note that the Debye–Waller factor is often ascribed to thermal motion, i.e., the are due to thermal motion, but any random displacements about a perfect lattice, not just thermal ones, will contribute to the Debye–Waller factor.
Disorder of the second kind
However, fluctuations that cause the correlations between pairs of atoms to decrease as their separation increases, causes the Bragg peaks in the structure factor of a crystal to broaden. To see how this works, we consider a one-dimensional toy model: a stack of plates with mean spacing . The derivation follows that in chapter 9 of Guinier's textbook.[6] This model has been pioneered by and applied to a number of materials by Hosemann and collaborators[7] over a number of years. Guinier and they termed this disorder of the second kind, and Hosemann in particular referred to this imperfect crystalline ordering as paracrystalline ordering. Disorder of the first kind is the source of the Debye–Waller factor.
To derive the model we start with the definition (in one dimension) of the

To start with we will consider, for simplicity an infinite crystal, i.e., . We will consider a finite crystal with disorder of the second-type below.
For our infinite crystal, we want to consider pairs of lattice sites. For large each plane of an infinite crystal, there are two neighbours planes away, so the above double sum becomes a single sum over pairs of neighbours either side of an atom, at positions and lattice spacings away, times . So, then



where is the probability density function for the separation of a pair of planes, lattice spacings apart. For the separation of neighbouring planes we assume for simplicity that the fluctuations around the mean neighbour spacing of a are Gaussian, i.e., that


![{\displaystyle p_{1}(\Delta x)={\frac {1}{\left(2\pi \sigma _{2}^{2}\right)^{1/2}}}\exp \left[-\left(\Delta x-a\right)^{2}/(2\sigma _{2}^{2})\right]}](../I/95f487305139e7ede9041c508aa8464a41365bb1.svg)
and we also assume that the fluctuations between a plane and its neighbour, and between this neighbour and the next plane, are independent. Then is just the convolution of two s, etc. As the convolution of two Gaussians is just another Gaussian, we have that


![{\displaystyle p_{m}(\Delta x)={\frac {1}{\left(2\pi m\sigma _{2}^{2}\right)^{1/2}}}\exp \left[-\left(\Delta x-ma\right)^{2}/(2m\sigma _{2}^{2})\right]}](../I/3ee45e99d1a13e4d17ebd8a6f2639b586d6b7934.svg)
The sum in is then just a sum of Fourier transforms of Gaussians, and so

for . The sum is just the real part of the sum and so the structure factor of the infinite but disordered crystal is
![{\displaystyle r=\exp[-q^{2}\sigma _{2}^{2}/2]}](../I/e5ffdc407cac909aa989eff76edb7b62a4030e30.svg)
![{\displaystyle \sum _{m=1}^{\infty }[r\exp(iqa)]^{m}}](../I/82729dc14555ad7f0af25342a751afbf3c43b598.svg)

This has peaks at maxima , where . These peaks have heights



i.e., the height of successive peaks drop off as the order of the peak (and so ) squared. Unlike finite-size effects that broaden peaks but do not decrease their height, disorder lowers peak heights. Note that here we assuming that the disorder is relatively weak, so that we still have relatively well defined peaks. This is the limit , where . In this limit, near a peak we can approximate , with and obtain




![{\displaystyle S(q)\approx {\frac {S(q_{P})}{1+{\frac {r}{(1-r)^{2}}}{\frac {\Delta q^{2}a^{2}}{2}}}}\approx {\frac {S(q_{P})}{1+{\frac {\Delta q^{2}}{[q_{P}^{2}\sigma _{2}^{2}/a]^{2}/2}}}}}](../I/d0d0f0ab7105dcad599811fcf8f4a71ce3e49282.svg)
which is a Lorentzian or Cauchy function, of FWHM , i.e., the FWHM increases as the square of the order of peak, and so as the square of the wave vector at the peak.

Finally, the product of the peak height and the FWHM is constant and equals , in the limit. For the first few peaks where is not large, this is just the limit.



Finite crystals with disorder of the second kind
For a one-dimensional crystal of size

where the factor in parentheses comes from the fact the sum is over nearest-neighbour pairs (), next nearest-neighbours (), ... and for a crystal of planes, there are pairs of nearest neighbours, pairs of next-nearest neighbours, etc.




Liquids
In contrast with crystals, liquids have no long-range order (in particular, there is no regular lattice), so the structure factor does not exhibit sharp peaks. They do however show a certain degree of short-range order, depending on their density and on the strength of the interaction between particles. Liquids are isotropic, so that, after the averaging operation in Equation (4), the structure factor only depends on the absolute magnitude of the scattering vector . For further evaluation, it is convenient to separate the diagonal terms in the double sum, whose phase is identically zero, and therefore each contribute a unit constant:


-
.
(9)

One can obtain an alternative expression for in terms of the radial distribution function :[8]

-
.
(10)

Ideal gas
In the limiting case of no interaction, the system is an ideal gas and the structure factor is completely featureless: , because there is no correlation between the positions and of different particles (they are independent random variables), so the off-diagonal terms in Equation (9) average to zero: .



![\langle \exp[-i{\mathbf {q}}({\mathbf {R}}_{j}-{\mathbf {R}}_{k})]\rangle =\langle \exp(-i{\mathbf {q}}{\mathbf {R}}_{j})\rangle \langle \exp(i{\mathbf {q}}{\mathbf {R}}_{k})\rangle =0](../I/03f0c0d741438cd1d54b86e1ab5d43498f96aca2.svg)
High-q limit
Even for interacting particles, at high scattering vector the structure factor goes to 1. This result follows from Equation (10), since is the Fourier transform of the "regular" function and thus goes to zero for high values of the argument . This reasoning does not hold for a perfect crystal, where the distribution function exhibits infinitely sharp peaks.

Low-q limit
In the low- limit, as the system is probed over large length scales, the structure factor contains thermodynamic information, being related to the isothermal compressibility of the liquid by the compressibility equation:

- .

Hard-sphere liquids
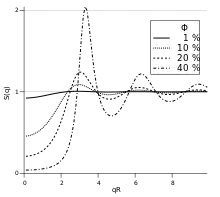

In the hard sphere model, the particles are described as impenetrable spheres with radius ; thus, their center-to-center distance and they experience no interaction beyond this distance. Their interaction potential can be written as:



This model has an analytical solution[9] in the Percus–Yevick approximation. Although highly simplified, it provides a good description for systems ranging from liquid metals[10] to colloidal suspensions.[11] In an illustration, the structure factor for a hard-sphere fluid is shown in the Figure, for volume fractions from 1% to 40%.
Polymers
In polymer systems, the general definition (4) holds; the elementary constituents are now the monomers making up the chains. However, the structure factor being a measure of the correlation between particle positions, one can reasonably expect that this correlation will be different for monomers belonging to the same chain or to different chains.
Let us assume that the volume contains identical molecules, each composed of monomers, such that ( is also known as the degree of polymerization). We can rewrite (4) as:



-
,
(11)

where indices label the different molecules and the different monomers along each molecule. On the right-hand side we separated intramolecular () and intermolecular () terms. Using the equivalence of the chains, (11) can be simplified:[12]




-
,
(12)

where is the single-chain structure factor.

Notes
- Warren, B. E. (1969). X-ray Diffraction. Addison Wesley.
- Cowley, J. M. (1992). Electron Diffraction Techniques Vol 1. Oxford Science. ISBN 9780198555582.
- Egami, T.; Billinge, S. J. L. (2012). Underneath the Bragg Peaks: Structural Analysis of Complex Material (2nd ed.). Elsevier. ISBN 9780080971339.
- "Structure Factor". Online Dictionary of CRYSTALLOGRAPHY. IUCr. Retrieved 15 September 2016.
- See Guinier, chapters 6-9
- Guinier, A (1963). X-Ray Diffraction. San Francisco and London: WH Freeman.
- Lindenmeyer, PH; Hosemann, R (1963). "Application of the Theory of Paracrystals to the Crystal Structure Analysis of Polyacrylonitrile". Journal of Applied Physics. 34 (1): 42. Bibcode:1963JAP....34...42L. doi:10.1063/1.1729086. Archived from the original on 2016-08-17.
- See Chandler, section 7.5.
- Wertheim, M. (1963). "Exact Solution of the Percus-Yevick Integral Equation for Hard Spheres". Physical Review Letters. 10 (8): 321–323. Bibcode:1963PhRvL..10..321W. doi:10.1103/PhysRevLett.10.321.
- Ashcroft, N.; Lekner, J. (1966). "Structure and Resistivity of Liquid Metals". Physical Review. 145 (1): 83–90. Bibcode:1966PhRv..145...83A. doi:10.1103/PhysRev.145.83.
- Pusey, P. N.; Van Megen, W. (1986). "Phase behaviour of concentrated suspensions of nearly hard colloidal spheres". Nature. 320 (6060): 340. Bibcode:1986Natur.320..340P. doi:10.1038/320340a0. S2CID 4366474.
- See Teraoka, Section 2.4.4.
References
- Als-Nielsen, N. and McMorrow, D. (2011). Elements of Modern X-ray Physics (2nd edition). John Wiley & Sons.
- Guinier, A. (1963). X-ray Diffraction. In Crystals, Imperfect Crystals, and Amorphous Bodies. W. H. Freeman and Co.
- Chandler, D. (1987). Introduction to Modern Statistical Mechanics. Oxford University Press.
- Hansen, J. P. and McDonald, I. R. (2005). Theory of Simple Liquids (3rd edition). Academic Press.
- Teraoka, I. (2002). Polymer Solutions: An Introduction to Physical Properties. John Wiley & Sons.
External links
- Structure Factor Tutorial located at the University of York.
- Definition of by IUCr
- Learning Crystallography, from the CSIC