Stopped-flow
Stopped-flow is an experimental technique for studying chemical reactions with a half time of the order of 1 ms, introduced by Britton Chance[1][2] and extended by Quentin Gibson[3] (Other techniques, such as the temperature-jump method,[4] are available for much faster processes.)
Description of the method
Summary
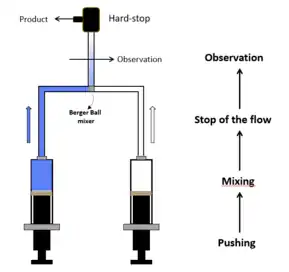
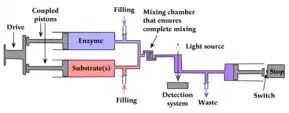
Stopped-flow spectrometry allows chemical kinetics of fast reactions (with half times of the order of milliseconds) to be studied in solution. It was first used primarily to study enzyme-catalyzed reactions. Then the stopped-flow rapidly found its place in almost all biochemistry, biophysics, and chemistry laboratories with a need to follow chemical reactions in the millisecond time scale. In its simplest form, a stopped-flow mixes two solutions. Small volumes of solutions are rapidly and continuously driven into a high-efficiency mixer. This mixing process then initiates an extremely fast reaction. The newly mixed solution travels to the observation cell and pushes out the contents of the cell (the solution remaining from the previous experiment or from necessary washing steps). The time required for this solution to pass from the mixing point to the observation point is known as dead time. The minimum injection volume will depend on the volume of the mixing cell. Once enough solution has been injected to completely remove the previous solution, the instrument reaches a stationary state and the flow can be stopped. Depending on the syringe drive technology, the flow stop is achieved by using a stop valve called the hard-stop or by using a stop syringe. The stopped-flow also sends a ‘start signal’ to the detector called the trigger so the reaction can be observed. The timing of the trigger is usually software controlled so the user can trigger at the same time the flow stops or a few milliseconds before the stop to check the stationary state has been reached.
So this is a very economical technique.
Reactant syringes
Two syringes are filled with solutions that do not undergo a chemical reaction until mixed together. These have pistons that are driven by a single drive piston or by independent stepping motors, so that they are coupled together and their contents are forced out simultaneously into a mixing device.
Mixing chamber
Once the two solutions are forced out of their syringes they enter a mixing system that has baffles to ensure complete mixing, with turbulent flow rather than laminar flow, which would allow the two solutions to flow side by side with incomplete mixing.
Dead time
The dead time is the time for the solutions to go from the mixing point to the observation point, it is the part of the kinetics which cannot be observed. So the lower the dead time, the more information the user can get. In older instruments this could be of the order of 1 ms, but improvements now allow a dead time of about 0.3 ms.[5]
Observation cell

The mixed reactants pass an observation cell that allows the reaction to be followed spectrophotometrically, typically by ultraviolet spectroscopy, fluorescence spectroscopy, circular dichroism or light scattering, and it is now common to combine several of these.[6] Observation cuvette with a short light path (0.75 to 1.5mm) are usually preferred for fluorescence measurements to reduce self-absorption effects. Observation cuvette with longer light path (0.5 cm to 1 cm) are preferred for absorbance measurements. Modern stopped-flow can accommodate different models of cells and it is possible to change the cuvette between two experiments. For stopped-flow X-ray measurements, a quartz capillary with thin wall is used to minimize quartz absorption. Simultaneous x-ray and absorbance measurements are possible in the same capillary.
Stopping
Once through the observation cell the mixture enters a third syringe that contains a piston that is driven by the flow to activate a switch to stop the flow and activate the observation.
Continuous flow
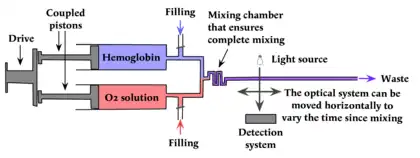
The stopped-flow method is a development of the continuous-flow method used by Hamilton Hartridge and Francis Roughton[7] to study the binding of O2 to hemoglobin. In the absence of any stopping system the reaction mixture passed to a long tube past an observation system (consisting in 1923 of a simple colorimeter) to waste. By moving the colorimeter along the tube, and knowing the flow rate, Hartridge and Roughton could measure the process after a known time.
In its time this was a revolutionary advance showing an apparently intractable problem (studying a process taking milliseconds with equipment that required seconds for each measurement) could be solved with simple equipment. However, in practice it was limited to reactants available in large quantities: for proteins this effectively limited it to reactions of hemoglobin. For practical purposes this approach is obsolete.
Quenched flow
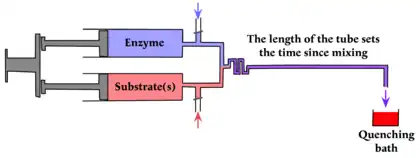
The stopped-flow method depends on the existence of spectroscopic properties that can be used for following the reaction. When that is not the case quenched flow provides an alternative that uses conventional chemical methods for analysis.[8] Instead of a mechanical stopping system the reaction is stopped by quenching, the products being delivered to a recipient that stops the reaction immediately, either by instantaneous freezing or by denaturing the enzyme with a chemical denaturant or exposing the sample to a denaturing light source. As in the continuous-flow method, the time between mixing and quenching can be varied by varying the length of the tube.
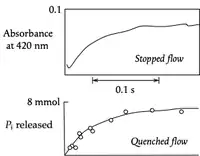
The pulsed quenched flow method introduced by Alan Fersht and Ross Jakes[9] overcomes the need for a long tube. The reaction is initiated exactly as in a stopped-flow experiment, but there is a third syringe that brings about quenching a definite and preset time after the initiation.
Quenched flow has both advantages and disadvantages with respect to stopped flow. On the one hand, chemical analysis makes it clear what process is being measured, whereas it may not always be obvious what process a spectroscopic signal represents. On the other hand, quenched flow is much more laborious, as each point along the time course must be determined separately. The image at left for catalysis by nitrogenase from Klebsiella pneumoniae[10] illustrates both of these points: the agreement in half times indicates that the absorbance at 420 nm measured the release of Pi, but the quenched-flow experiment required 11 data points.
References
- Chance, Britton (1951). "Rapid and Sensitive Spectrophotometry. I. The Accelerated and Stopped‐Flow Methods for the Measurement of the Reaction Kinetics and Spectra of Unstable Compounds in the Visible Region of the Spectrum". Review of Scientific Instruments. 22 (8): 619–627. doi:10.1063/1.1746019.
- Chance, Britton; Legallais, Victor (1951). "Rapid and Sensitive Spectrophotometry. II. A Stopped‐Flow Attachment for a Stabilized Quartz Spectrophotometer". Review of Scientific Instruments. 22 (8): 627–634. doi:10.1063/1.1746020.
- Gibson, Q. H. (1954). "Stopped-flow apparatus for the study of rapid reactions". Discussions of the Faraday Society. 17: 137. doi:10.1039/df9541700137.
- Eigen, M. (1954). "Methods for investigation of ionic reactions in aqueous solutions with half-times as short as 10–9sec. Application to neutralization and hydrolysis reactions". Discuss. Faraday Soc. 17: 194–205. doi:10.1039/df9541700194.
- Clark, Charles R. (1997). "A Stopped-Flow Kinetics Experiment for Advanced Undergraduate Laboratories: Formation of Iron(III) Thiocyanate". Journal of Chemical Education. 74 (10): 1214. Bibcode:1997JChEd..74.1214C. doi:10.1021/ed074p1214.
- Guillerm, Jessica; Frère, Jean-Marie; Meersman, Filip; Matagne, André (2021). "The right-handed parallel β-helix topology of Erwinia chrysanthemi pectin methylesterase is intimately associated with both sequential folding and resistance to high pressure". Biomolecules. 11: 1083. doi:10.3390/biom11081083. PMC 8392785. PMID 34439750.
- Hartridge, H.; Roughton, F. J. W. (1923). "A method for measuring the velocity of very rapid chemical reactions". Proceedings of the Royal Society A. 104 (726): 376–394. Bibcode:1923RSPSA.104..376H. doi:10.1098/rspa.1923.0116.
- Pinsent, B R W (1954). "A quenching method for studying rapid reactions". Discussions of the Faraday Society. 17: 140–141. doi:10.1039/df9541700140.
- Fersht, A R; Jakes, R (1975). "Demonstration of two reaction pathways for aminoacylation of transfer-RNA: application of pulsed quenched flow technique". Biochemistry. 14 (15): 3350–3356. doi:10.1021/bi00686a010.
- Thorneley, R N F; Cornish-Bowden, A (1977). "Kinetics of nitrogenase of Klebsiella-pneumoniae: heterotropic interactions between magnesium-adenosine 5'-diphosphate and magnesium-adenosine 5'-triphosphate". Biochem. J. 165 (2): 255–262. doi:10.1042/bj1650255. PMC 1164896.
Further reading
- Alan Fersht (1998). Structure and Mechanism in Protein Science: Guide to Enzyme Catalysis and Protein Folding. New York: W. H. Freeman. pp. 132–168. ISBN 9780716732686.
- Athel Cornish-Bowden (2012). Fundamentals of Enzyme Kinetics (4th ed.). Weinheim: Wiley-Blackwell. pp. 391–396. ISBN 978-3527330744.