Bond order potential
Bond order potential is a class of empirical (analytical) interatomic potentials which is used in molecular dynamics and molecular statics simulations. Examples include the Tersoff potential,[1] the EDIP potential, the Brenner potential,[2] the Finnis–Sinclair potentials, [3] ReaxFF,[4] and the second-moment tight-binding potentials.[5] They have the advantage over conventional molecular mechanics force fields in that they can, with the same parameters, describe several different bonding states of an atom, and thus to some extent may be able to describe chemical reactions correctly. The potentials were developed partly independently of each other, but share the common idea that the strength of a chemical bond depends on the bonding environment, including the number of bonds and possibly also angles and bond lengths. It is based on the Linus Pauling bond order concept [1] [6] and can be written in the form

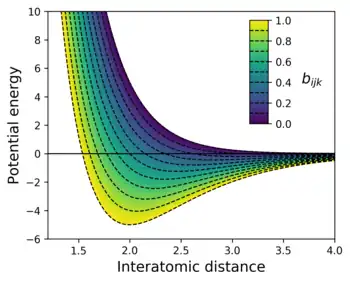
This means that the potential is written as a simple pair potential depending on the distance between two atoms , but the strength of this bond is modified by the environment of the atom via the bond order . is a function that in Tersoff-type potentials depends inversely on the number of bonds to the atom , the bond angles between sets of three atoms , and optionally on the relative bond lengths , .[1] In case of only one atomic bond (like in a diatomic molecule), which corresponds to the strongest and shortest possible bond. The other limiting case, for increasingly many number of bonds within some interaction range, and the potential turns completely repulsive (as illustrated in the figure to the right).







Alternatively, the potential energy can be written in the embedded atom model form

where is the electron density at the location of atom . These two forms for the energy can be shown to be equivalent (in the special case that the bond-order function contains no angular dependence).[7]

A more detailed summary of how the bond order concept can be motivated by the second-moment approximation of tight binding and both of these functional forms derived from it can be found in.[8]
The original bond order potential concept has been developed further to include distinct bond orders for sigma bonds and pi bonds in the so-called BOP potentials.[9]
Extending the analytical expression for the bond order of the sigma bonds to include fourth moments of the exact tight binding bond order reveals contributions from both sigma- and pi- bond integrals between neighboring atoms. These pi-bond contributions to the sigma bond order are responsible to stabilize the asymmetric before the symmetric (2x1) dimerized reconstruction of the Si(100) surface.[10]
Also the ReaxFF potential can be considered a bond order potential, although the motivation of its bond order terms is different from that described here.
References
- Tersoff, J. (1988). "New empirical approach for the structure and energy of covalent systems". Phys. Rev. B. 37 (12): 6991–7000. Bibcode:1988PhRvB..37.6991T. doi:10.1103/PhysRevB.37.6991. PMID 9943969.
- Brenner, D. W. (1990). "Empirical potential for hydrocarbons for use in simulating the chemical vapor deposition of diamond films". Phys. Rev. B. 42 (15): 9458–9471. Bibcode:1990PhRvB..42.9458B. doi:10.1103/PhysRevB.42.9458. PMID 9995183. Archived from the original on September 22, 2017.
- Finnis, M. W. (1984). "A simple empirical N-body potential for transition metals". Philos. Mag. A. 50 (1): 45–55. Bibcode:1984PMagA..50...45F. doi:10.1080/01418618408244210.
- ReaxFF: A Reactive Force Field for Hydrocarbons, Adri C. T. van Duin, Siddharth Dasgupta, Francois Lorant, and William A. Goddard III, J. Phys. Chem. A, 2001, 105 (41), pp 9396–9409
- Cleri, F.; V. Rosato (1993). "Tight-binding potentials for transition metals and alloys". Phys. Rev. B. 48 (1): 22–33. Bibcode:1993PhRvB..48...22C. doi:10.1103/PhysRevB.48.22. PMID 10006745.
- Abell, G. C. (1985). "Empirical chemical pseudopotential theory of molecular and metallic bonding". Phys. Rev. B. 31 (10): 6184–6196. Bibcode:1985PhRvB..31.6184A. doi:10.1103/PhysRevB.31.6184. PMID 9935490.
- Brenner, D. (1989). "Relationship between the embedded-atom method and Tersoff potentials". Phys. Rev. Lett. 63 (9): 1022. Bibcode:1989PhRvL..63.1022B. doi:10.1103/PhysRevLett.63.1022. PMID 10041250.
- Albe, K.; K. Nordlund (2002). "Modeling the metal-semiconductor interaction: Analytical bond-order potential for platinum-carbon". Phys. Rev. B. 65 (19): 195124. Bibcode:2002PhRvB..65s5124A. doi:10.1103/physrevb.65.195124.
- Pettifor, D. G.; I. I. Oleinik (1999). "Analytic bond-order potentials beyond Tersoff-Brenner. I. Theory". Phys. Rev. B. 59 (13): 8487–8499. Bibcode:1999PhRvB..59.8487P. doi:10.1103/PhysRevB.59.8487.
- Kuhlmann, V.; K. Scheerschmidt (2007). "σ-bond expression for an analytic bond-order potential: Including π and on-site terms in the fourth moment". Phys. Rev. B. 76 (1): 014306. Bibcode:2007PhRvB..76a4306K. doi:10.1103/PhysRevB.76.014306.