3C syndrome
3C syndrome is a rare condition whose symptoms include heart defects, cerebellar hypoplasia, and cranial dysmorphism. It was first described in the medical literature in 1987 by Ritscher and Schinzel, for whom the disorder is sometimes named.
| 3C syndrome | |
|---|---|
| Other names | CCC dysplasia, Craniocerebellocardiac dysplasia[1] or Ritscher–Schinzel syndrome,[2] |
| Specialty | Medical genetics |
Signs and symptoms
The classical triad of symptoms that defines 3C syndrome includes certain heart defects, hypoplasia (underdevelopment) of the cerebellum, and cranial dysmorphisms, which can take various forms. The heart defects and cranial dysmorphisms are heterogeneous in individuals who are all classed as having Ritscher-Schinzel syndrome.[2]
Heart defects commonly seen with Ritscher-Schinzel syndrome are associated with the endocardial cushion and are the most important factor in determining a diagnosis. The mitral valve and tricuspid valve of the heart can be malformed, the atrioventricular canal can be complete instead of developing into the interatrial septum and interventricular septum, and conotruncal heart defects, which include tetralogy of Fallot, double outlet right ventricle, transposition of the great vessels,[2] and hypoplastic left heart syndrome. Aortic stenosis and pulmonary stenosis have also been associated with 3C syndrome.[3]
The cranial dysmorphisms associated with 3C syndrome are heterogeneous and include a degree of macrocephaly, a large anterior fontanel, a particularly prominent occiput and forehead, ocular hypertelorism (wide-set eyes), slanted palpebral fissures, cleft palate, a depressed nasal bridge, cleft palate with associated bifid uvula,[2] low-set ears, micrognathia (an abnormally small jaw),[4] brachycephaly (flattened head), and ocular coloboma.[3] Low-set ears are the most common cranial dysmorphism seen in 3C syndrome, and ocular coloboma is the least common of the non-concurrent symptoms (cleft lip co-occurring with cleft palate is the least common).[5]
Cranial dysplasias associated with 3C syndrome are also reflected in the brain. Besides the cerebellar hypoplasia, cysts are commonly found in the posterior cranial fossa, the ventricles and the cisterna magna are dilated/enlarged, and Dandy–Walker malformation is present. These are reflected in the developmental delays typical of the disease.[4][5] 75% of children with 3C syndrome have Dandy-Walker malformation and hydrocephalus.[6]
Signs and symptoms in other body systems are also associated with 3C syndrome. In the skeletal system, ribs may be absent, and hemivertebrae, syndactyly (fusion of fingers together), and clinodactyly (curvature of the fifth finger) may be present.[3][6] In the GI and genitourinary systems, anal atresia, hypospadia (misplaced urethra), and hydronephrosis may exist. Adrenal hypoplasia and growth hormone deficiency are associated endocrine consequences of Ritscher-Schinzel syndrome.[3] Some immunodeficiency has also been reported in connection with 3C syndrome.[6] Many children with the disorder die as infants due to severe congenital heart disease.[4] The proband of Ritscher and Schinzel's original study was still alive at the age of 21.[7] A fetus with 3C syndrome may have an umbilical cord with one umbilical artery instead of two.[3][7]
Genetics
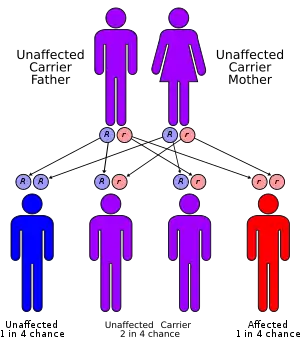
3C syndrome is an autosomal recessive disease, caused by a mutation on the long arm of chromosome 8 at 8q24.13, the locus for KIAA0196,[4] the gene for the protein strumpellin. Strumpellin is highly expressed in skeletal muscle cells and mutations in it are also associated with spastic paraplegia. Strumpellin is involved in endosomal transport and cell death processes.[8] The mutation occurs at a splice site and causes a substantial decrease in the amount of strumpellin produced by the cell. The phenotype is similar to 6pter-p24 deletion syndrome and 6p25 deletion syndrome but has a different etiology.[4][7]
- Screening for the condition prenatally may be done with ultrasound.
- First-trimester ultrasounds can detected nuchal abnormalities
- Second-trimester ultrasounds can pick up characteristic major structural abnormalities.[9]
- Prenatal diagnosis is possible through genetic testing.
- Chorionic villus sampling or chorionic villus biopsy (CVS) in the first-trimester.
- Amniocentesis in the second-trimester.
Because 3C syndrome is an autosomal recessive disorder, parents with one child with the disorder have a 25% chance of having another child with the disorder.[4]
Diagnosis
Differential diagnosis
There is an overlap in symptoms between 3C syndrome and Joubert syndrome. Joubert syndrome often manifests with similar cerebellar hypoplasia and its sequelae, including hyperpnea, ataxia, changes in eye movement, and cleft lip and palate. Occasionally, Joubert syndrome will include heart malformations. Brachmann–de Lange syndrome must also be differentiated from 3C syndrome. It presents with similar craniofacial and heart abnormalities and can include Dandy–Walker phenotype, making it difficult to distinguish. Dandy-Walker malformation is also occasionally seen in Ellis–Van Creveld syndrome, which is characterized by heart defects and malformed alveolar ridge.[5] Many disorders include the Dandy–Walker phenotype and thus it is not pathognomonic for 3C syndrome.[10]
CHARGE syndrome can also be misdiagnosed. This is because both CHARGE syndrome and 3C syndrome share symptoms of ocular colobomas, cardiac defects, growth retardation, and minor facial abnormalities.[2]
Coffin–Siris syndrome presents with fifth-finger deformities and congenital heart defects. It is distinguished from 3C syndrome by differences in facial dysmorphisms.[6]
Management
The outcome of this disease is dependent on the severity of the cardiac defects. Approximately 1 in 3 children with this diagnosis require shunting for the hydrocephaly that is often a consequence. Some children require extra assistance or therapy for delayed psychomotor and speech development, including hypotonia.[2]
Prognosis
Prognoses for 3C syndrome vary widely based on the specific constellation of symptoms seen in an individual. Typically, the gravity of the prognosis correlates with the severity of the cardiac abnormalities. For children with less severe cardiac abnormalities, the developmental prognosis depends on the cerebellar abnormalities that are present. Severe cerebellar hypoplasia is associated with growth and speech delays, as well as hypotonia and general growth deficiencies.[5]
Epidemiology
3C syndrome is very rare, occurring in less than 1 birth per million.[2] Because of consanguinity due to a founder effect, it is much more common in a remote First Nations village in Manitoba, where 1 in 9 people carries the recessive gene.[4]
History
The syndrome was first reported in 1987 in two sisters who had similar craniofacial abnormalities, Dandy–Walker phenotype, and congenital heart abnormalities. Neither of the parents was affected, indicating that the disorder was transmitted in an autosomal recessive pattern.[4][11] The syndrome's symptoms were further refined in 1989 when the third case of the syndrome was reported, with similar craniofacial abnormalities to the first two cases, ventricular septal defect, and enlargement of the cisterna magna and fourth ventricle of the brain.[5]
Other animals
Animal models of 3C syndrome have not been created; however, strumpellin is a highly conserved protein, with 12 known homologs and 83 known orthologs.[8]
References
- Disease ID 5666 at NIH's Office of Rare Diseases
- "3C syndrome". Orphanet. Retrieved 11 April 2014.
- Kniffin, Cassandra L.; Jackson, John F. (6 January 2014). "Ritscher-Schinzel Syndrome - Clinical Synopsis". Online Mendelian Inheritance in Man. Johns Hopkins University. Retrieved 12 April 2014.
- Kniffin, Cassandra L.; McCusick, Victor A. (6 January 2014). "Ritscher-Schinzel Syndrome". Online Mendelian Inheritance in Man. Johns Hopkins University. Retrieved 11 April 2014.
- Leonardi, Michael L.; Pai, G. Shashidhar; Wilkes, Beth; Lebel, Robert Roger (15 August 2001). "Ritscher-Schinzel cranio-cerebello-cardiac (3C) syndrome: Report of four new cases and review". American Journal of Medical Genetics. 102 (3): 237–242. doi:10.1002/ajmg.1449. PMID 11484200.
- Gorlin, Robert J.; Cohen Jr., Michael; Hennekam, Raoul C.M. (2001). Syndromes of the Head and Neck (4 ed.). Oxford University Press. ISBN 9780199747726.
- Jones, Kenneth Lyons; Jones, Marilyn Crandall; del Campo, Miguel (2013). Smith's Recognizable Patterns of Human Malformation (7th ed.). Elsevier Health Sciences. ISBN 9780323186681.
- "KIAA0196". NIH. Retrieved 12 April 2014.
- Rusnak, A. J., Hadfield, M. I., Chudley, A. E., Marles, S. L., Reid, G. J., & Chodirker, B. N. (2008). Increased Nuchal Translucency Thickness: A Potential Indicator for Ritscher-Schinzel Syndrome. Fetal Diagnosis & Therapy, 24(4), 395-399. doi:10.1159/000165697
- Albright, A. Leland; Pollack, Ian F. (2011). Principles and Practice of Pediatric Neurosurgery. Thieme. ISBN 9781604064605.
- Ritscher, D.; Schinzel, A.; Boltshauser, E.; Briner, J.; Arbenz, U.; Sigg, P. (February 1987). "Dandy-Walker(like) malformation, atrio-ventricular septal defect and a similar pattern of minor anomalies in 2 sisters: a new syndrome?". American Journal of Medical Genetics. 26 (2): 481–491. doi:10.1002/ajmg.1320260227. PMID 3812597.