Acyl-CoA dehydrogenase
Acyl-CoA dehydrogenases (ACADs) are a class of enzymes that function to catalyze the initial step in each cycle of fatty acid β-oxidation in the mitochondria of cells. Their action results in the introduction of a trans double-bond between C2 (α) and C3 (β) of the acyl-CoA thioester substrate.[1] Flavin adenine dinucleotide (FAD) is a required co-factor in addition to the presence of an active site glutamate in order for the enzyme to function.
The following reaction is the oxidation of the fatty acid by FAD to afford an α,β-unsaturated fatty acid thioester of Coenzyme A:

ACADs can be categorized into three distinct groups based on their specificity for short-, medium-, or long-chain fatty acid acyl-CoA substrates. While different dehydrogenases target fatty acids of varying chain length, all types of ACADs are mechanistically similar. Differences in the enzyme occur based on the location of the active site along the amino acid sequence.[2]
ACADs are an important class of enzymes in mammalian cells because of their role in metabolizing fatty acids present in ingested food materials. This enzyme's action represents the first step in fatty acid metabolism (the process of breaking long chains of fatty acids into acetyl CoA molecules). Deficiencies in these enzymes are linked to genetic disorders involving fatty acid oxidation (i.e. metabolic disorders).[3]
ACAD enzymes have been identified in animals (of which there are 9 major eukaryotic classes), as well as plants,[4] nematodes,[5] fungi,[6] and bacteria.[7] Five of these nine classes are involved in fatty acid β-oxidation (SCAD, MCAD, LCAD, VLCAD, and VLCAD2), and the other four are involved in branched chain amino acid metabolism (i3VD, i2VD, GD, and iBD). Most acyl-CoA dehydrogenases are α4 homotetramers, and in two cases (for very long chain fatty acid substrates) they are α2 homodimers. An additional class of acyl-CoA dehydrogenase was discovered that catalyzes α,β-unsaturation reactions with steroid-CoA thioesters in certain types of bacteria.[8][9] This class of ACAD was demonstrated to form α2β2 heterotetramers, rather than the usual α4 homotetramer, a protein architecture that evolved in order to accommodate a much larger steroid-CoA substrate.[10][11]
ACADs are classified as EC 1.3.99.3.
Structure
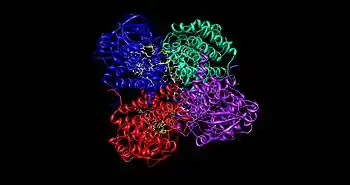
The medium chain acyl-CoA dehydrogenase (MCAD) is the best known structure of all ACADs, and is the most commonly deficient enzyme within the class that leads to metabolic disorders in animals.[1] This protein is a homotetramer with each subunit containing roughly 400 amino acids and one equivalent of FAD per monomer. The tetramer is classified as a "dimer of dimers" with an overall diameter of approximately 90 Å.[2]
The interface between the two monomers of a single dimer of an ACAD contains the FAD binding sites and has extensive bonding interactions. In contrast, the interface between the two dimers has fewer interactions. There are a total of 4 active sites within the tetramer, each of which contains a single FAD molecule and an acyl-CoA substrate binding site. This gives a total of four FAD molecules and four acyl-CoA substrate binding sites per enzyme.
FAD is bound between the three domains of the monomer, where only the nucleotide portion is accessible. FAD binding contributes significantly to overall enzyme stability. The acyl-CoA substrate is bound completely within each monomer of the enzyme. The active site is lined with the residues F252, T255, V259, T96, T99, A100, L103, Y375, Y375, and E376. The area of interest within the substrate becomes wedged between Glu 376 and FAD, lining up the molecules into an ideal position for the reaction.[1]
MCAD can bind to a rather broad range of chain-lengths in the acyl-CoA substrate, however studies show that its specificity tends to target octanoyl-CoA (C8-CoA).[12]
A novel ACAD enzyme architecture in some species of steroid-utilizing bacteria (Actinomycetota and Pseudomonadota) was discovered, and is involved in the utilization of ubiquitous steroid substrates like cholesterol by pathogenic organisms like Mycobacterium tuberculosis. Genetically, the structure is encoded by two separate genes (open reading frames) that form an obligate α2β2 heterotetramic complex. The structure was most likely the result of an evolutionary event that caused gene duplication and partial loss of function, since half of the FAD cofactor binding residues are in each gene, and only make a complete binding site when expressed together as a complex. This probably allowed for the substrate binding site to open up considerably to accommodate much larger polycyclic-CoA substrates, rather than fatty acids of varying chain lengths.
Mechanism
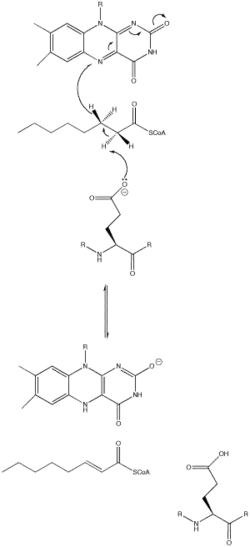
The acyl-CoA dehydrogenase mechanism proceeds through an E2 elimination. This elimination is initiated by a glutamate residue, which, while necessary for the mechanism, is not conserved.[1]
The residue appears in a wide range of locations within the different types of the enzyme (it is Glu 376 in MCAD). The glutamate residue deprotonates the pro-R hydrogen of the alpha carbon. Hydrogen bonding of the substrate's carbonyl oxygen to both the 2'-OH of the ribityl side-chain of FAD and to the main chain N-H of the previously mentioned glutamate residue lowers the pKa of this proton, allowing it to be readily removed by glutamate.[1]
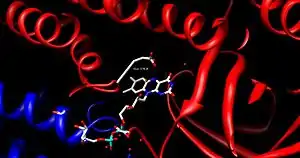
As the alpha carbon is being deprotonated, the pro-R hydrogen of the beta carbon leaves as a hydride to FAD in a concerted step. It adds to the Re face of FAD at the N-5 position, and the enzyme holds FAD in place through hydrogen bonding with the pyrimidine portion and hydrophobic interactions with the dimethylbenzene portion. The substrate has now been transformed into an α,β unsaturated thioester.[1]
As FAD picks up the hydride, the carbonyl oxygen adjacent to the N-1 nitrogen becomes negatively charged. These electrons are in resonance with the N-1 nitrogen, distributing and stabilizing the resulting negative charge. The charge is also stabilized by hydrogen bonding between the oxygen and nitrogen of interest and various residues within the enzyme.[1]
Clinical significance
Deficiencies in acyl-CoA dehydrogenases result in decreased ability to oxidize fatty acids, thereby signifying metabolic dysfunction. Medium-chain acyl-CoA dehydrogenase deficiencies (MCADD) are well known and characterized because they occur most commonly among acyl-CoA dehydrogenases, leading to fatty acid oxidation disorders and the potential of life-threatening metabolic diseases. Some symptoms of medium-chain acyl-CoA dehydrogenase deficiency include intolerance to fasting, hypoglycemia, and sudden infant death syndrome. These symptoms are seen as directly connected to the inability to metabolize fats. Intolerance to fasting and hypoglycemia result from the inability to gain energy and make sugar from fat stores, which is how most of humans' excess energy is stored. Also, fatty acids can begin to accumulate in the blood, lowering the blood's pH and causing acidosis.[1]
MCAD is related to / has an association with sudden infant death. Approximately 90% of cases of MCAD are due to a single point mutation where the lysine at position 304 (Lys304)is replaced by a glutamate residue and this prevents the enzyme from properly functioning.[1] It is reported that, every year, 1 in 20,000 infants is born with a deficiency in his/her medium-chain acyl-CoA dehydrogenases that is caused by a mutation. The mutation is recessive, and often parents of children who have the deficiency can be diagnosed afterward as carriers.[3]
In humans the most common naturally occurring mutation in MCAD is located at amino acid residue Lys-304.[1] The altered residue occurs as a result of a single-point mutation in which the lysine side chain is replaced by a glutamate. Lys-304 typically interacts with surrounding amino acid residues by forming hydrogen bonds with Gln-342, Asp-300, and Asp-346. When a mutation causes glutamate to take the place of lysine, an additional negative charge is introduced at that site, which disrupts the normally occurring H-bonding. Such a disruption alters the folding pattern of the enzyme, ultimately compromising its stability and inhibiting its function in fatty acid oxidation.[12] The efficiency of the mutated protein is about 10 times lower than that of the natural protein.[13] This can lead to the symptoms of the deficiency listed above.
See also
- Acyl CoA
- Beta oxidation
References
- Thorpe, C.; Kim, J. J. (June 1995). "Structure and Mechanism of Action of the Acyl-CoA Dehydrogenases". FASEB J. 9 (9): 718–25. doi:10.1096/fasebj.9.9.7601336. PMID 7601336. S2CID 42549744.
- Kim JJ, Wang M, Paschke R (August 1993). "Crystal structures of medium-chain acyl-CoA dehydrogenase from pig liver mitochondria with and without substrate". Proc. Natl. Acad. Sci. U.S.A. 90 (16): 7523–7. Bibcode:1993PNAS...90.7523K. doi:10.1073/pnas.90.16.7523. PMC 47174. PMID 8356049.
- Touma EH, Charpentier C (January 1992). "Medium chain acyl-CoA dehydrogenase deficiency". Arch. Dis. Child. 67 (1): 142–5. doi:10.1136/adc.67.1.142. PMC 1793557. PMID 1739332.
- Bode, K.; Hooks, M.A.; Couee, I. (1999). "Identification, separation, and characterization of acyl-coenzyme A dehydrogenases involved in mitochondrial β-oxidation in higher plants". Plant Physiol. 119 (4): 1305–1314. doi:10.1104/pp.119.4.1305. PMC 32015. PMID 10198089.
- Komuniecki, R.; Fekete, S.; Thissen-Parra, J. (1985). "Purification and characterization of the 2‐methyl branched‐chain Acyl-CoA dehydrogenase, an enzyme involved in NADH-dependent enoyl-CoA reduction in anaerobic mitochondria of the nematode, Ascaris suum". J Biol Chem. 260 (8): 4770–4777. doi:10.1016/S0021-9258(18)89138-4. PMID 3988734.
- Kionka, C.; Kunau, W.H. (1985). "Inducible β-oxidation pathway in Neurospora crassa". J Bacteriol. 161 (1): 153–157. doi:10.1128/jb.161.1.153-157.1985. PMC 214849. PMID 3155714.
- Campbell, J.W.; Cronan, J.E. Jr. (2002). "The enigmatic Escherichia coli fadE gene is yafH". J. Bacteriol. 184 (13): 3759–64. CiteSeerX 10.1.1.333.9931. doi:10.1128/JB.184.13.3759-3764.2002. PMC 135136. PMID 12057976.
- Thomas, S.T.; Sampson, N.S. (2013). "Mycobacterium tuberculosis utilizes a unique heterotetrameric structure for dehydrogenation of the cholesterol side chain". Biochemistry. 52 (17): 2895–2904. doi:10.1021/bi4002979. PMC 3726044. PMID 23560677.
- Wipperman, M.F.; Yang, M.; Thomas, S.T.; Sampson, N.S. (2013). "Shrinking the FadE Proteome of Mycobacterium tuberculosis: Insights into Cholesterol Metabolism through Identification of an α2β2 Heterotetrameric Acyl Coenzyme A Dehydrogenase Family". J. Bacteriol. 195 (19): 4331–4341. doi:10.1128/JB.00502-13. PMC 3807453. PMID 23836861.
- Voskuil, M.I. (2013). "Mycobacterium tuberculosis Cholesterol Catabolism Requires a New Class of Acyl Coenzyme A Dehydrogenase". J. Bacteriol. 195 (19): 4319–4321. doi:10.1128/JB.00867-13. PMC 3807469. PMID 23893117.
- Wipperman, Matthew, F.; Thomas, Suzanne, T.; Sampson, Nicole, S. (2014). "Pathogen roid rage: Cholesterol utilization by Mycobacterium tuberculosis". Crit. Rev. Biochem. Mol. Biol. 49 (4): 269–93. doi:10.3109/10409238.2014.895700. PMC 4255906. PMID 24611808.
- Kieweg V, Kräutle FG, Nandy A, et al. (June 1997). "Biochemical characterization of purified, human recombinant Lys304→Glu medium-chain acyl-CoA dehydrogenase containing the common disease-causing mutation and comparison with the normal enzyme" (PDF). Eur. J. Biochem. 246 (2): 548–56. doi:10.1111/j.1432-1033.1997.00548.x. PMID 9208949.
- Nasser I, Mohsen AW, Jelesarov I, Vockley J, Macheroux P, Ghisla S (September 2004). "Thermal unfolding of medium-chain acyl-CoA dehydrogenase and iso(3)valeryl-CoA dehydrogenase: study of the effect of genetic defects on enzyme stability". Biochim. Biophys. Acta. 1690 (1): 22–32. doi:10.1016/j.bbadis.2004.04.008. PMID 15337167.
- "Molecular graphics images were produced using the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIH P41 RR-01081). "
- Pettersen EF, Goddard TD, Huang CC, et al. (October 2004). "UCSF Chimera—a visualization system for exploratory research and analysis". J Comput Chem. 25 (13): 1605–12. CiteSeerX 10.1.1.456.9442. doi:10.1002/jcc.20084. PMID 15264254. S2CID 8747218.
- Lee HJ, Wang M, Paschke R, Nandy A, Ghisla S, Kim JJ (September 1996). "Crystal structures of the wild type and the Glu376Gly/Thr255Glu mutant of human medium-chain acyl-CoA dehydrogenase: influence of the location of the catalytic base on substrate specificity". Biochemistry. 35 (38): 12412–20. doi:10.1021/bi9607867. PMID 8823176.
Further reading
- Wenz A, Thorpe C, Ghisla S (October 1981). "Inactivation of general acyl-CoA dehydrogenase from pig kidney by a metabolite of hypoglycin A". J. Biol. Chem. 256 (19): 9809–12. doi:10.1016/S0021-9258(19)68697-7. PMID 7275979.
- Engst S, Vock P, Wang M, Kim JJ, Ghisla S (January 1999). "Mechanism of activation of acyl-CoA substrates by medium chain acyl-CoA dehydrogenase: interaction of the thioester carbonyl with the flavin adenine dinucleotide ribityl side chain". Biochemistry. 38 (1): 257–67. doi:10.1021/bi9815041. PMID 9890906.
- Voskuil, M.I. (2013). "Mycobacterium tuberculosis Cholesterol Catabolism Requires a New Class of Acyl Coenzyme A Dehydrogenase". J. Bacteriol. 195 (19): 4319–4321. doi:10.1128/JB.00867-13. PMC 3807469. PMID 23893117.
External links
- Acyl-CoA+Dehydrogenase at the US National Library of Medicine Medical Subject Headings (MeSH)