Carnitine
Carnitine is a quaternary ammonium compound involved in metabolism in most mammals, plants, and some bacteria.[1][2][3][4] In support of energy metabolism, carnitine transports long-chain fatty acids into mitochondria to be oxidized for energy production, and also participates in removing products of metabolism from cells.[3] Given its key metabolic roles, carnitine is concentrated in tissues like skeletal and cardiac muscle that metabolize fatty acids as an energy source.[3] Generally individuals, including strict vegetarians, synthesize enough L-carnitine in vivo.[1]
 | |
 | |
| Clinical data | |
|---|---|
| AHFS/Drugs.com | Micromedex Detailed Consumer Information |
| Routes of administration | Oral, intravenous |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | <10% |
| Protein binding | None |
| Metabolism | slightly |
| Excretion | Urine (>95%) |
| Identifiers | |
IUPAC name
| |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII |
|
| KEGG | |
| ChEBI | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.006.343 |
| Chemical and physical data | |
| Formula | C7H15NO3 |
| Molar mass | 161.201 g·mol−1 |
| 3D model (JSmol) | |
SMILES
| |
InChI
| |
| | |
Carnitine exists as one of two stereoisomers (the two enantiomers d-carnitine (S-(+)-) and l-carnitine (R-(−)-)).[5] Both are biologically active, but only l-carnitine naturally occurs in animals, and d-carnitine is toxic as it inhibits the activity of the l-form.[6] At room temperature, pure carnitine is a white powder, and a water-soluble zwitterion with low toxicity. Derived from amino acids,[7] carnitine was first extracted from meat extracts in 1905, leading to its name from Latin, "caro/carnis" or flesh.[2]
Some individuals with genetic or medical disorders (such as preterm infants) cannot make enough carnitine, requiring dietary supplementation.[1][3][4] Despite common carnitine supplement consumption among athletes for improved exercise performance or recovery, there is insufficient high-quality clinical evidence to indicate it provides any benefit.[3][4]
Biosynthesis and metabolism
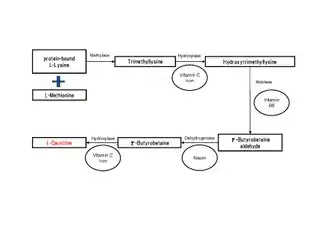
Many eukaryotes have the ability to synthesize carnitine, including humans.[1][3] Humans synthesize carnitine from the substrate TML (6-N-trimethyllysine), which is in turn derived from the methylation of the amino acid lysine.[1] TML is then hydroxylated into hydroxytrimethyllysine (HTML) by trimethyllysine dioxygenase (TMLD), requiring the presence of ascorbic acid and iron. HTML is then cleaved by HTML aldolase (a pyridoxal phosphate requiring enzyme), yielding 4-trimethylaminobutyraldehyde (TMABA) and glycine. TMABA is then dehydrogenated into gamma-butyrobetaine in an NAD+-dependent reaction, catalyzed by TMABA dehydrogenase.[1] Gamma-butyrobetaine is then hydroxylated by gamma butyrobetaine hydroxylase (a zinc binding enzyme[8]) into l-carnitine, requiring iron in the form of Fe2+.[1][9]
Carnitine is involved in transporting fatty acids across the mitochondrial membrane, by forming a long chain acetylcarnitine ester and being transported by carnitine palmitoyltransferase I and carnitine palmitoyltransferase II.[10] Carnitine also plays a role in stabilizing Acetyl-CoA and coenzyme A levels through the ability to receive or give an acetyl group.[1]
Tissue distribution of carnitine-biosynthetic enzymes
The tissue distribution of carnitine-biosynthetic enzymes in humans indicates TMLD to be active in the liver, heart, muscle, brain and highest in the kidneys.[1] HTMLA activity is found primarily in the liver. The rate of TMABA oxidation is greatest in the liver, with considerable activity also in the kidneys.[1]
Carnitine shuttle system
The free-floating fatty acids, released from adipose tissues to the blood, bind to carrier protein molecule known as serum albumin that carry the fatty acids to the cytoplasm of target cells such as the heart, skeletal muscle, and other tissue cells, where they are used for fuel. But before the target cells can use the fatty acids for ATP production and β oxidation, the fatty acids with chain lengths of 14 or more carbons must be activated and subsequently transported into mitochondrial matrix of the cells in three enzymatic reactions of the carnitine shuttle.[11]
The first reaction of the carnitine shuttle is a two-step process catalyzed by a family of isozymes of acyl-CoA synthetase that are found in the outer mitochondrial membrane, where they promote the activation of fatty acids by forming a thioester bond between the fatty acid carboxyl group and the thiol group of coenzyme A to yield a fatty acyl–CoA.[11]
In the first step of the reaction, acyl-CoA synthetase catalyzes the transfer of adenosine monophosphate group (AMP) from an ATP molecule onto the fatty acid generating a fatty acyl–adenylate intermediate and a pyrophosphate group (PPi). The pyrophosphate, formed from the hydrolysis of the two high-energy bonds in ATP, is immediately hydrolyzed to two molecules of Pi by inorganic pyrophosphatase. This reaction is highly exergonic which drives the activation reaction forward and makes it more favorable. In the second step, the thiol group of a cytosolic coenzyme A attacks the acyl-adenylate, displacing AMP to form thioester fatty acyl-CoA.[11]
In the second reaction, acyl-CoA is transiently attached to the hydroxyl group of carnitine to form fatty acylcarnitine. This transesterification is catalyzed by an enzyme found in the outer membrane of the mitochondria known as carnitine acyltransferase 1 (also called carnitine palmitoyltransferase 1, CPT1).[11]
The fatty acylcarnitine ester formed then diffuses across the intermembrane space and enters the matrix by facilitated diffusion through carnitine-acylcarnitine translocase (CACT) located on the inner mitochondrial membrane. This antiporter returns one molecule of carnitine from the matrix to the intermembrane space for every one molecule of fatty acyl–carnitine that moves into the matrix.[11]
In the third and final reaction of the carnitine shuttle, the fatty acyl group is transferred from fatty acyl-carnitine to coenzyme A, regenerating fatty acyl–CoA and a free carnitine molecule. This reaction takes place in the mitochondrial matrix and is catalyzed by carnitine acyltransferase 2 (also called carnitine palmitoyltransferase 2, CPT2), which is located on the inner face of the inner mitochondrial membrane. The carnitine molecule formed is then shuttled back into the intermembrane space by the same cotransporter (CACT) while the fatty acyl-CoA enters β-oxidation.[11]
Regulation of fatty acid β oxidation
The carnitine-mediated entry process is a rate-limiting factor for fatty acid oxidation and is an important point of regulation.[11]
Inhibition
The liver starts actively making triglycerides from excess glucose when it is supplied with glucose that cannot be oxidized or stored as glycogen. This increases the concentration of malonyl-CoA, the first intermediate in fatty acid synthesis, leading to the inhibition of carnitine acyltransferase 1, thereby preventing fatty acid entry into the mitochondrial matrix for β oxidation. This inhibition prevents fatty acid breakdown while synthesis occurs.[11]
Activation
Carnitine shuttle activation occurs due to a need for fatty acid oxidation which is required for energy production. During vigorous muscle contraction or during fasting, ATP concentration decreases and AMP concentration increases leading to the activation of AMP-activated protein kinase (AMPK). AMPK phosphorylates acetyl-CoA carboxylase, which normally catalyzes malonyl-CoA synthesis. This phosphorylation inhibits acetyl-CoA carboxylase, which in turn lowers the concentration of malonyl-CoA. Lower levels of malonyl-CoA disinhibit carnitine acyltransferase 1, allowing fatty acid import to the mitochondria, ultimately replenishing the supply of ATP.[11]
Transcription factors
Peroxisome proliferator-activated receptor alpha (PPARα) is a nuclear receptor that functions as a transcription factor. It acts in muscle, adipose tissue, and liver to turn on a set of genes essential for fatty acid oxidation, including the fatty acid transporters carnitine acyltransferases 1 and 2, the fatty acyl–CoA dehydrogenases for short, medium, long, and very long acyl chains, and related enzymes.[11]
PPARα functions as a transcription factor in two cases; as mentioned before when there is an increased demand for energy from fat catabolism, such as during a fast between meals or long-term starvation. Besides that, the transition from fetal to neonatal metabolism in the heart. In the fetus, fuel sources in the heart muscle are glucose and lactate, but in the neonatal heart, fatty acids are the main fuel that require the PPARα to be activated so it is able in turn to activate the genes essential for fatty acid metabolism in this stage.[11]
Metabolic defects of fatty acid oxidation
More than 20 human genetic defects in fatty acid transport or oxidation have been identified. In case of fatty acid oxidation defects, acyl-carnitines accumulate in mitochondria and are transferred into the cytosol, and then into the blood. Plasma levels of acylcarnitine in newborn infants can be detected in a small blood sample by tandem mass spectrometry.[11]
When β oxidation is defective because of either mutation or deficiency in carnitine, the ω (omega) oxidation of fatty acids becomes more important in mammals. Actually, the ω Oxidation of Fatty Acids is another pathway for F-A degradation in some species of vertebrates and mammals that occurs in the endoplasmic reticulum of the liver and kidney, it is the oxidation of the ω carbon—the carbon most far from the carboxyl group (in contrast to oxidation which occurs at the carboxyl end of fatty acid, in the mitochondria).[1][11]

Physiological effects
As an example of normal synthesis, a 70 kilograms (150 lb) person would produce 11–34 mg of carnitine per day.[1] Adults eating mixed diets of red meat and other animal products ingest some 60–180 mg of carnitine per day, while vegans consume about 10–12 mg per day.[3] Most (54–86%) carnitine obtained from the diet is absorbed in the small intestine before entering the blood.[3] The total body content of carnitine is about 20 grams (0.71 oz) in a person weighing 70 kilograms (150 lb), with nearly all of it contained within skeletal muscle cells.[3] Carnitine metabolizes at rates of about 400 μmol (65mg) per day, an amount less than 1% of total body stores.[1]
Deficiency
Carnitine deficiency is rare in healthy people without metabolic disorders, indicating that most people have normal, adequate levels of carnitine normally produced through fatty acid metabolism.[1] One study found that vegans showed no signs of carnitine deficiency.[1] Infants, especially premature infants, have low stores of carnitine, necessitating use of carnitine-fortified infant formulas as a replacement for breast milk, if necessary.[1]
Two types of carnitine deficiency states exist. Primary carnitine deficiency is a genetic disorder of the cellular carnitine-transporter system that typically appears by the age of five with symptoms of cardiomyopathy, skeletal-muscle weakness, and hypoglycemia.[1][3] Secondary carnitine deficiencies may happen as the result of certain disorders, such as chronic kidney failure, or under conditions that reduce carnitine absorption or increase its excretion, such as the use of antibiotics, malnutrition, and poor absorption following digestion.[1][3]
Supplementation
Despite widespread interest among athletes to use carnitine for improvement of exercise performance, inhibit muscle cramps, or enhance recovery from physical training, the quality of research for these possible benefits has been low, prohibiting any conclusion of effect.[1][3] At supplement amounts of 2–6 grams (0.071–0.212 oz) per day over a month, there was no consistent evidence that carnitine affected exercise or physical performance.[3] Carnitine supplements do not improve oxygen consumption or metabolic functions when exercising, nor do they increase the amount of carnitine in muscle.[1][3] There is no evidence that L-carnitine influences fat metabolism or aids in weight loss.[3]
Male fertility
The carnitine content of seminal fluid is directly related to sperm count and motility, suggesting that the compound might be of value in treating male infertility.[1]
Diseases
Carnitine has been studied in various cardiometabolic conditions, indicating it is under preliminary research for its potential as an adjunct in heart disease and diabetes, among numerous other disorders.[1] Carnitine has no effect on preventing all-cause mortality associated with cardiovascular diseases,[12] and has no significant effect on blood lipids.[1][13]
Although there is some evidence from meta-analyses that L-carnitine supplementation improved cardiac function in people with heart failure, there is insufficient research to determine its overall efficacy in lowering the risk or treating cardiovascular diseases.[1][12]
There is only preliminary clinical research to indicate the use of L-carnitine supplementation for improving symptoms of type 2 diabetes, such as improving glucose tolerance or lowering fasting levels of blood glucose.[1][14]
The kidneys contribute to overall homeostasis in the body, including carnitine levels. In the case of renal impairment, urinary elimination of carnitine increasing, endogenous synthesis decreasing, and poor nutrition as a result of disease-induced anorexia can result in carnitine deficiency.[1] Carnitine has no effect on most parameters in end-stage kidney disease, although it may lower C-reactive protein, a biomarker for systemic inflammation.[15] Carnitine blood levels and muscle stores can become low, which may contribute to anemia, muscle weakness, fatigue, altered levels of blood fats, and heart disorders.[1] Some studies have shown that supplementation of high doses of l-carnitine (often injected) may aid in anemia management.[1]
Sources
The form present in the body is l-carnitine, which is also the form present in food. Food sources rich in l-carnitine are animal products, particularly beef and pork.[1] Red meats tend to have higher levels of l-carnitine.[1][13] Adults eating diverse diets that contain animal products attain about 23-135 mg of carnitine per day.[1][16] Vegans get noticeably less (about 10–12 mg) since their diets lack these carnitine-rich animal-derived foods. Approximately 54% to 86% of dietary carnitine is absorbed in the small intestine, then enters the blood.[1] Even carnitine-poor diets have little effect on total carnitine content, as the kidneys conserve carnitine.[13]
| Food | Milligrams (mg) |
|---|---|
| Beef steak, cooked, 4 ounces (110 g) | 56–162 |
| Ground beef, cooked, 4 ounces (110 g) | 87–99 |
| Milk, whole, 1 cup (237 g) | 8 |
| Codfish, cooked, 4 ounces (110 g) | 4–7 |
| Chicken breast, cooked, 4 ounces (110 g) | 3–5 |
| Ice cream, ½ cup (125 mL) | 3 |
| Cheese, cheddar, 2 ounces (57 g) | 2 |
| Whole–wheat bread, 2 slices | 0.2 |
| Asparagus, cooked, ½ cup (62 g) | 0.1 |
In general, omnivorous humans each day consume between 2 and 12 µmol kg−1 of body weight, accounting for 75% of carnitine in the body. Humans endogenously produce 1.2 µmol kg−1 of body weight of carnitine on a daily basis, accounting for 25% of the carnitine in the body.[1][3] Strict vegetarians obtain little carnitine from dietary sources (0.1 µmol kg−1 of body weight daily), as it is mainly found in animal-derived foods.[1][17]
L-Carnitine, acetyl-l-carnitine, and propionyl-l-carnitine are available in dietary supplement pills or powders, with a daily amount of 0.5 to 1 g considered to be safe.[1][3] It is also a drug approved by the Food and Drug Administration to treat primary and certain secondary carnitine-deficiency syndromes secondary to inherited diseases.[1][3]
Drug interactions and adverse effects
Carnitine interacts with pivalate-conjugated antibiotics such as pivampicillin. Chronic administration of these antibiotics increases the excretion of pivaloyl-carnitine, which can lead to carnitine depletion.[1] Treatment with the anticonvulsants valproic acid, phenobarbital, phenytoin, or carbamazepine significantly reduces blood levels of carnitine.[4]
When taken in the amount of roughly 3 grams (0.11 oz) per day, carnitine may cause nausea, vomiting, abdominal cramps, diarrhea, and body odor smelling like fish.[1][4] Other possible adverse effects include skin rash, muscle weakness, or seizures in people with epilepsy.[4]
History
Levocarnitine was approved by the U.S. Food and Drug Administration as a new molecular entity under the brand name Carnitor on December 27, 1985.[4][5]
See also
- Acetylcarnitine
- Gamma-butyrobetaine dioxygenase
- Glycine Propionyl-l-Carnitine (GPLC)
- Meldonium
- Systemic primary carnitine deficiency
References
- "L-Carnitine". Micronutrient Information Center, Linus Pauling Institute, Oregon State University, Corvallis, OR. 2019-12-01. Retrieved 2020-04-29.
- Bremer J (October 1983). "Carnitine--metabolism and functions". Physiological Reviews. 63 (4): 1420–80. doi:10.1152/physrev.1983.63.4.1420. PMID 6361812.
- "Carnitine". Office of Dietary Supplements, US National Institutes of Health. 2017-10-10. Retrieved 2020-04-29.
- "L-carnitine: Uses, benefits and dosage". Drugs.com. 2020-01-20. Retrieved 2020-04-29.
- "Levocarnitine". PubChem, National Library of Medicine, US National Institutes of Health. 2020-04-25. Retrieved 2020-04-29.
- Matsuoka M, Igisu H (July 1993). "Comparison of the effects of L-carnitine, D-carnitine and acetyl-L-carnitine on the neurotoxicity of ammonia". Biochemical Pharmacology. 46 (1): 159–64. doi:10.1016/0006-2952(93)90360-9. PMID 8347126.
- Cox RA, Hoppel CL (December 1973). "Biosynthesis of carnitine and 4-N-trimethylaminobutyrate from 6-N-trimethyl-lysine". The Biochemical Journal. 136 (4): 1083–90. doi:10.1042/bj1361083. PMC 1166060. PMID 4786530.
- Tars K, Rumnieks J, Zeltins A, Kazaks A, Kotelovica S, Leonciks A, et al. (August 2010). "Crystal structure of human gamma-butyrobetaine hydroxylase". Biochemical and Biophysical Research Communications. 398 (4): 634–9. doi:10.1016/j.bbrc.2010.06.121. PMID 20599753.
- Strijbis K, Vaz FM, Distel B (May 2010). "Enzymology of the carnitine biosynthesis pathway". IUBMB Life. 62 (5): 357–62. doi:10.1002/iub.323. PMID 20306513.
- Flanagan JL, Simmons PA, Vehige J, Willcox MD, Garrett Q (April 2010). "Role of carnitine in disease". Nutrition & Metabolism. 7: 30. doi:10.1186/1743-7075-7-30. PMC 2861661. PMID 20398344.
- Nelson DL, Cox MM, Lehninger AL (2017). Lehninger principles of biochemistry (7th ed.). New York, NY: W.H. Freeman and Company.
- Shang R, Sun Z, Li H (July 2014). "Effective dosing of ʟ-carnitine in the secondary prevention of cardiovascular disease: a systematic review and meta-analysis". BMC Cardiovascular Disorders. 14: 88. doi:10.1186/1471-2261-14-88. PMC 4223629. PMID 25044037.
- Huang H, Song L, Zhang H, Zhang H, Zhang J, Zhao W (1 January 2013). "Influence of ʟ-carnitine supplementation on serum lipid profile in hemodialysis patients: a systematic review and meta-analysis". Kidney & Blood Pressure Research. 38 (1): 31–41. doi:10.1159/000355751. PMID 24525835.
- Bene J, Hadzsiev K, Melegh B (March 2018). "Role of carnitine and its derivatives in the development and management of type 2 diabetes". Nutrition & Diabetes. 8 (1): 8. doi:10.1038/s41387-018-0017-1. PMC 5856836. PMID 29549241.
- Chen Y, Abbate M, Tang L, Cai G, Gong Z, Wei R, Zhou J, Chen X (February 2014). "ʟ-Carnitine supplementation for adults with end-stage kidney disease requiring maintenance hemodialysis: a systematic review and meta-analysis". The American Journal of Clinical Nutrition. 99 (2): 408–22. doi:10.3945/ajcn.113.062802. PMID 24368434.
- Rebouche, C. J. (2004). "Kinetics, pharmacokinetics, and regulation of ʟ-carnitine and acetyl-ʟ-carnitine metabolism". Annals of the New York Academy of Sciences. 1033 (1): 30–41. Bibcode:2004NYASA1033...30R. doi:10.1196/annals.1320.003. PMID 15591001. S2CID 24803029.
- Lombard KA, Olson AL, Nelson SE, Rebouche CJ (August 1989). "Carnitine status of lactoovovegetarians and strict vegetarian adults and children". The American Journal of Clinical Nutrition. 50 (2): 301–6. doi:10.1093/ajcn/50.2.301. PMID 2756917.