Central nervous system primitive neuroectodermal tumor
A central nervous system primitive neuroectodermal tumor, often abbreviated as PNET, supratentorial PNET, or CNS-PNET,[1] is one of the 3 types of embryonal central nervous system tumors defined by the World Health Organization (medulloblastoma, atypical teratoid rhabdoid tumor, and PNET).[2] It is considered an embryonal tumor because it arises from cells partially differentiated or still undifferentiated from birth.[1] Those cells are usually neuroepithelial cells,[1][2][3] stem cells destined to turn into glia or neurons.[4] It can occur anywhere within the spinal cord and cerebrum and can have multiple sites of origins, with a high probability of metastasis through cerebrospinal fluid (CSF).[1][2]
| Central nervous system primitive neuroectodermal tumor | |
|---|---|
 | |
| Primitive neuroectodermal tumor of the central nervous system in a 5-year-old |
PNET has five subtypes of tumors: neuroblastoma, ganglioneuroblastoma, medulloepithelioma, ependymoblastoma, and not otherwise specified PNET.[1] It is similar to medulloblastoma regarding histology but different regarding genetic factors and tumor site. It is a rare disease occurring mostly among children,[1][2] accounting for 1.9 to 7% of childhood brain tumors.[2] Symptoms involve emotional, visual, motor, and speech defects.[2] Magnetic resonance imaging (MRI) and computed tomography (CT) are used to diagnose PNETs.[2] Even though a universal treatment plan hasn't been stablished yet, common strategies involve chemotherapy and radiotherapy for individuals older than 3 years of age.[1][2] Their efficacy, however, is still controversial.[2] Surgery can be used to remove mass affected by tumorous cells.[2] The prognosis of the disease is more positive for adults than for children, who have a higher probability of having sequelae from the tumor.[1][2]
It is important to note that this classification term has been removed from the latest WHO classification of CNS tumors as of 2016. Instead PNETs are now included into the category of "Embryonal Tumors with Multilayered Rosettes" along with ependymoblastoma and embryonal tumor with abundant neuropil and true rosettes (ETANTR).[5]
Classification
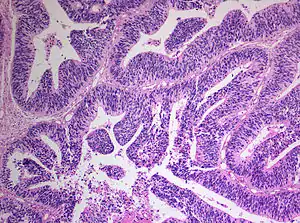
The World Health Organization has classified the central nervous system primitive neuroectodermal tumors into five subtypes: neuroblastoma, ganglioneuroblastoma, medulloepithelioma, ependymoblastoma, and not otherwise specified PNET.[1] The last one encompasses the PNETs with varying characteristics that hasn't been well defined yet.[1] Neuroblastomas are PNETS that involve the process of cell differentiation into neurons,[1][2] while ganglioneuroblastomas are PNETs that involve ganglion cells.[1]
Medulloepithelioma, on the other hand, are tumors involving the constant cell division on the epithelium tissue where bundle of neuron endings are located.[1] Such tissue will differentiate into a similar form as the embryonic neural tube, also known as the starting structure of the central nervous system.[1][2][3] Medulloepitheliomas also present a pattern known as rosettes, characterized by the arrangement of a bundle of cells into circular shapes and around a center or a neuropil.[1] Ependymoblastoma also present rosettes as well as a higher density of cells.[1][3] It involves the process of differentiation into ependymal cells.[2][3]

Further classification types have come up but not yet approved by the World Health Organization.[1] The term "embryonal tumor with abundant neuropil and true rosettes", or ETANTR, has been proposed as a sixth subtype of PNET.[1] However, the still unofficial term "embryonal tumor with multilayered rosettes" (ETMR) has been more frequently used and encompasses ETANTRs, medulloepitheliomas, ependymoblastomas, and variants of PNETs with presence of rosettes and with no well defined classification.[3]
PNET vs. medulloblastoma
The differentiation between primitive neuroectodermal tumor in the central nervous system and medulloblastoma is recent.[1][2] According to the World Health Organization, both tumors have the same histology but primitive neuroectodermal tumors occur outside the cerebellum.[2] Moreover, it has been documented that both have different genetic expression and mutations.[1][2] Another essential difference between them is the location of their respective blood vessels within the brain.[2] It has also been theorized that PNETs influence mainly glia cells while medulloblastomas influence mainly neural behavior, however such theory hasn't been confirmed yet.[1] Medulloblastomas are more frequent than PNETs, representing 10% of all child deaths caused by cancer.[2] They also present better prognosis: children affected by medulloblastoma reach the 5 year survival mark in 70-80% of cases, while children affected by PNET reach the 5 year survival mark in less than 50% of cases.[1]
Risk factors
The rate of PNETs in not correlated with sex, but it shows a correlation with age.[1] Most cases occur in children around 5 years of age, having a very low frequency in adults.[1] Regarding genetic mutations, a specific type of gene alteration that directly leads to this tumor hasn't been defined yet.[1] However, a positive correlation between individuals with Li-Fraumeni syndrome with a mutation in the gene p53 and PNET has been reported.[2] A significant number of individuals with mutations on the rb tumor suppressor gene have also developed the tumor.[2] Such gene encodes for the protein Rb responsible for stopping the cell cycle at the G1 phase.[6] Another possible contributing factor are mutations in the CREB-binding protein, whose function includes activating transcription,[6] but this interaction still need to be studied further.[2] It has also been presumed that the tumor can arise from cranial irradiation.[2]
Diagnosis
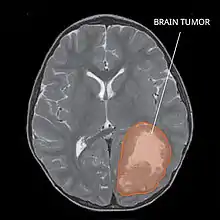
Most children that develop primitive neuroectodermal tumors are diagnosed early in life, usually at around 3–6.8 years of age.[2] Symptoms patients present at time of diagnosis include irritable mood, visual difficulties, lethargy, and ataxia.[2] The circumference of the patient's head might also become enlarged and they might be subject to seizures, especially if they have less than one year of life.[2]
Several analysis can be used to determine the presence of the disease. Physical examinations showing papilledema, visual field defects, cranial nerves palsy, dysphasia, and focal neurological deficits are evidences for possible tumor.[2] PNETs can also be spotted through computed tomography (CT) and magnetic resonance imaging (MRI).[2] In images produced by MRIs, an irregular augmentation among a solid mass will indicated the presence of tumor.[3] However, the results of MRIs are usually ambiguous in defining the presence for this specific tumor.[2] In CT scans, the presence of PNETs will be indicated by an elevated density and an increase in volume of the brain.[2] The CT scan can also show calcification,[3] which is present in 41-44% of PNET cases.[2] Since the tumor can be replicated in other parts of the nervous system through the cerebrospinal fluid (CSF), a CSF analysis can also be conducted.[2] A spinal MRI is a fourth type of analysis that is useful in investigating the level of tumor propagation to the spinal cord.[2]
Treatment
There is not a standardized procedure to treat primitive neuroectodermal tumors.[2] Common strategies involve risk-adapted radiotherapy combined with chemotherapy and stem cell rescue.[1] For patients younger than 2–3 years of age, treatment with radiation is not used, once they are in a more vulnerable phase and, thus, more prone to risks in development.[1] Examinations such as CSF analysis and spinal MRIs are used to investigate the effectiveness of treatment in preventing metastasis.[2]
A method for eliminating tumorous mass is surgery, where the best outcome would be total resection, meaning the complete removal of the tumor.[2] Along with the surgery, several measures that contribute to a safe procedure can be taken: urine exams, transfusion, and the constant supervision of arterial pressure.[2] Possible problems that arise from the surgery include hemorrhage, brain edema, and hemiparesis.[2] MRIs are typically done after 1 or 2 days of postoperative in order to inspect the amount of tumor remaining.[2]
Prognosis
The probability of primitive neuroectodermal tumors to have recurrence and metastasize through cerebrospinal fluid is relatively high.[3] The outcome of PNET is more positive when the individual is an adult, independent of age subgroups, or an older child.[2] Less than 50% of children survive more than 5 years,[1] while the majority of adults live to 7 years.[2] The reason the prognosis for such tumor is worst in children is due to the higher probability of the tumor spreading to the rest of the nervous system through the cerebrospinal fluid and growing again.[2] Moreover, children have the probability of developing deficiencies in cognitive processes, problems in the endocrine system, and psychological obstacles after the disease.[2] Adults, on the other hand, don't show such propensity.[2] As a consequence, 37.7% of children affected by the tumor live to 4 years.[2]
The effect of treatment strategies such as chemotherapy and radiation therapy on the prognosis of the disease is still controversial, with studies claiming either their benefits or their ineffectiveness.[2] The same holds true for the relationship between volume of tumor removed by surgery and survival.[2] Furthermore, factors such as tumor size, location of origin, race, and sex of individual don't show any influence on the outcome of the disease.[2] However, interactions of some factors such as tumor site, age, and treatment strategy can affect one's prognosis.[2] For instance, when younger children below the age of three develop tumors originating in places other than the pineal gland are treated with chemotherapy, they present better outcomes than those with pineal tumors and treated with chemotherapy.[2]
References
- Karajannis, Matthias A.; Zagzag, David, eds. (2015). "Molecular Pathology of Nervous System Tumors". Molecular Pathology Library. 8. doi:10.1007/978-1-4939-1830-0. ISBN 978-1-4939-1829-4. ISSN 1935-987X.
- Hayat, M.A., ed. (2014). "Tumors of the Central Nervous System, Volume 13". Tumors of the Central Nervous System. 13. doi:10.1007/978-94-007-7602-9. ISBN 978-94-007-7601-2. ISSN 2215-096X.
- Fuller, Christine E. (2009-10-23), "Oligodendroglial Tumors", Atlas of Pediatric Brain Tumors, Springer New York, pp. 39–46, doi:10.1007/978-1-4419-1062-2_4, ISBN 9781441910615
- Nelesen, Richard A (March 2000). "Biological Psychology: An Introduction to Behavioral, Cognitive, and Clinical Neuroscience, 2nd edition. Mark R. Rosenweig, Arnold L. Leiman, and S. Marc Breedlove, Sinauer Associates, Inc., Sunderland MA, 1999. 561+92 pp. ISBN 0-87893-791-9". Biological Psychology. 52 (2): 185–186. doi:10.1016/s0301-0511(99)00025-3. ISSN 0301-0511. S2CID 54349873.
- Louis DN, Ohgaki H, Wiestler OD, Cavenee WK "WHO Classification of Tumours of the Central Nervous System. 4th Edition Revised"
- Baker, Henry V (June 2003). "Essential Genetics: A Genomics Perspective. Third Edition. By Daniel L Hartl and , Elizabeth W Jones. Sudbury (Massachusetts): Jones and Bartlett Publishers. $78.95 (paper). xxvi + 613 p; ill.; index. 2002". The Quarterly Review of Biology. 78 (2): 225–226. doi:10.1086/377959. ISBN 0-7637-1852-1. ISSN 0033-5770.