Medulloblastoma
Medulloblastoma is a common type of primary brain cancer in children. It originates in the part of the brain that is towards the back and the bottom, on the floor of the skull, in the cerebellum, or posterior fossa.[1]
| Medulloblastoma | |
|---|---|
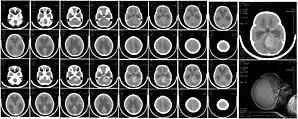 | |
| CT scan, showing a tumorous mass in the posterior fossa, giving rise to obstructive hydrocephalus, in a six-year-old girl | |
| Pronunciation |
|
| Specialty | Neuro-oncology, neurosurgery |
The brain is divided into two main parts, the larger cerebrum on top and the smaller cerebellum below towards the back. They are separated by a membrane called the tentorium. Tumors that originate in the cerebellum or the surrounding region below the tentorium are, therefore, called infratentorial.
Historically medulloblastomas have been classified as a primitive neuroectodermal tumor (PNET), but it is now known that medulloblastoma is distinct from supratentorial PNETs and they are no longer considered similar entities.[2]
Medulloblastomas are invasive, rapidly growing tumors that, unlike most brain tumors, spread through the cerebrospinal fluid and frequently metastasize to different locations along the surface of the brain and spinal cord. Metastasis all the way down to the cauda equina at the base of the spinal cord is termed "drop metastasis".
The cumulative relative survival rate for all age groups and histology follow-up was 60%, 52%, and 47% at 5 years, 10 years, and 20 years, respectively, with children doing better than adults.[3]
Signs and symptoms
Signs and symptoms are mainly due to secondary increased intracranial pressure due to blockage of the fourth ventricle and tumors are usually present for 1 to 5 months before diagnosis is made. The child typically becomes listless, with repeated episodes of vomiting, and a morning headache, which may lead to a misdiagnosis of gastrointestinal disease or migraine.[4] Soon after, the child will develop a stumbling gait, truncal ataxia, frequent falls, diplopia, papilledema, and sixth cranial nerve palsy. Positional vertigo and nystagmus are also frequent, and facial sensory loss or motor weakness may be present. Decerebrate attacks appear late in the disease.
Extraneural metastasis to the rest of the body is rare, and when it occurs, it is in the setting of relapse, more commonly in the era prior to routine chemotherapy.
Pathogenesis
Medulloblastomas are usually found in the vicinity of the fourth ventricle, between the brainstem and the cerebellum. Tumors with similar appearance and characteristics originate in other parts of the brain, but they are not identical to medulloblastoma.[5]
Although medulloblastomas are thought to originate from immature or embryonal cells at their earliest stage of development, the cell of origin depends on the subgroup of medulloblastoma. WNT tumors originate from the lower rhombic lip of the brainstem, while SHH tumors originate from the external granular layer.[6]
Currently, medulloblastomas are thought to arise from cerebellar stem cells that have been prevented from dividing and differentiating into their normal cell types. This accounts for the histologic variants seen on biopsy. Both perivascular pseudorosette and Homer Wright pseudorosette formations are highly characteristic of medulloblastomas and are seen in up to half of cases.[7] The classic rosette with tumor cells around a central lumen can be seen.[8]
In the past, medulloblastoma was classified using histology, but recent integrated genomic studies have revealed that medulloblastoma is composed of four distinct molecular and clinical variants termed WNT/β-catenin, Sonic Hedgehog, Group 3, and Group 4.[9] Of these subgroups, WNT patients have an excellent prognosis and group 3 patients have a poor prognosis. Also, a subgroup-specific alternative splicing further confirms the existence of distinct subgroups and highlights the transcriptional heterogeneity between subgroups.[10] Amplification of the Sonic Hedgehog pathway is the best characterized subgroup, with 25% of human tumors having mutations in Patched, Sufu (Suppressor of Fused Homolog), Smoothened, or other genes in this pathway.[11][12] Medulloblastomas are also seen in Gorlin syndrome as well as Turcot syndrome. Recurrent mutations in the genes CTNNB1, PTCH1, MLL2, SMARCA4, DDX3X, CTDNEP1, KDM6A, and TBR1 were identified in individuals with medulloblastoma.[13] Additional pathways disrupted in some medulloblastomas include MYC, Notch, BMP, and TGF-β signaling pathways.[11][12][4][14][15][16][17][1]
Diagnosis
The tumor is distinctive on T1- and T2-weighted MRI with heterogeneous enhancement and a typical location adjacent to and extension into the fourth ventricle. Histologically, the tumor is solid, pink-gray in color, and is well circumscribed. The tumor is very cellular, with high mitotic activity, little cytoplasm, and a tendency to form clusters and rosettes.
The Chang staging system can be used in making the diagnosis .
Correct diagnosis of medulloblastoma may require ruling out atypical teratoid rhabdoid tumor.[18]
_in_adult.JPG.webp) Cerebellar medulloblastoma in an adult
Cerebellar medulloblastoma in an adult_in_adult.JPG.webp) Cerebellar medulloblastoma in an adult
Cerebellar medulloblastoma in an adult
Treatment
Treatment begins with maximal surgical removal of the tumor. The addition of radiation to the entire neuraxis and chemotherapy may increase the disease-free survival. Some evidence indicates that proton beam irradiation reduces the impact of radiation on the cochlear and cardiovascular areas and reduces the cognitive late effects of cranial irradiation.[19][20] This combination may permit a 5-year survival in more than 80% of cases. The presence of desmoplastic features such as connective tissue formation offers a better prognosis. Prognosis is worse if the child is less than 3 years old, degree of resection is inadequate, or if any CSF, spinal, supratentorial, or systemic spread occurs. Dementia after radiotherapy and chemotherapy is a common outcome appearing two to four years following treatment. Side effects from radiation treatment can include cognitive impairment, psychiatric illness, bone growth retardation, hearing loss, and endocrine disruption.[1][4][14] Increased intracranial pressure may be controlled with corticosteroids or a ventriculoperitoneal shunt. A new approach to monitor tumor development and treatment response by liquid biopsy is promising, but remains challenging.[21]
Chemotherapy
Chemotherapy is often used as part of treatment. Evidence of benefit, however, is not clear as of 2013.[22] A few different chemotherapeutic regimens for medulloblastoma are used; most involve a combination of lomustine, cisplatin, carboplatin, vincristine, or cyclophosphamide. In younger patients (less than 3–4 years of age), chemotherapy can delay, or in some cases possibly even eliminate, the need for radiotherapy. However, both chemotherapy and radiotherapy often have long-term toxicity effects, including delays in physical and cognitive development, higher risk of second cancers, and increased cardiac disease risks.[23][24]
Outcomes
Array-based karyotyping of 260 medulloblastomas resulted in the following clinical subgroups based on cytogenetic profiles:[25]
- Poor prognosis: gain of 6q or amplification of MYC or MYCN
- Intermediate: gain of 17q or an i(17q) without gain of 6q or amplification of MYC or MYCN
- Excellent prognosis: 6q and 17q balanced or 6q deletion
Transcriptional profiling shows the existence of four main subgroups (Wnt, Shh, Group 3, and Group 4).[9]
- Very good prognosis: WNT group, CTNNB1 mutation
- Infants good prognosis, others intermediate: SHH group, PTCH1/SMO/SUFU mutation, GLI2 amplification, or MYCN amplification
- Poor prognosis: Group 3, MYC amplification, photoreceptor/GABAergic gene expression
- Intermediate prognosis: Group 4, gene expression of neuronal/glutamatergic, CDK6 amplification, MYCN amplification
Survival
The historical cumulative relative survival rate for all age groups and histology follow-up was 60%, 52%, and 47% at 5 years, 10 years, and 20 years, respectively. Patients diagnosed with a medulloblastoma or PNET are 50 times more likely to die than a matched member of the general population. The most recent population-based (SEER) 5-year relative survival rates are 69% overall: 72% in children (1–9 years) and 67% in adults (20+ years). The 20-year survival rate is 51% in children. Children and adults have different survival profiles, with adults faring worse than children only after the fourth year after diagnosis (after controlling for increased background mortality). Before the fourth year, survival probabilities are nearly identical.[3] Long-term sequelae of standard treatment include hypothalamic-pituitary and thyroid dysfunction and intellectual impairment. The hormonal and intellectual deficits created by these therapies causes significant impairment of the survivors.[26]
In current clinical studies, the patients are divided into low-, standard- and high-risk groups:
- Depending on the study, healing rates of up to 100% are achieved in the low-risk group (usually WNT-activated).[27] The current efforts are therefore moving in the direction of reducing the intensity of the therapy and thus the negative long-term consequences while confirming the high healing rates.[28]
- In the HIT-SIOP PNET 4 study, in which 340 children and adolescents of the standard-risk group between the ages of four and 21 from several European countries participated, the 5-year survival rate was between 85% and 87% depending on the randomization. Around 78% of the patients remained without relapse for 5 years and are therefore considered to be cured.[29] After a relapse, the prognosis was very poor. Despite intensive treatment, only four of 66 patients were still alive 5 years after a relapse.[30]
- A US study involved 161 patients between the ages of three and 21 with a high-risk profile. Depending on the randomization, half of the patients additionally received carboplatin daily during the radiation. The 5-year survival rate of patients with carboplatin was 82%, those without 68%.[31] The European SIOP PNET 5 study is currently taking place and will run until April 2024, in which an attempt is made to confirm the promising results with carboplatin during irradiation in the standard risk group.[28]
Epidemiology
Medulloblastomas affect just under two people per million per year, and affect children 10 times more than adults.[32] Medulloblastoma is the second-most frequent brain tumor in children after pilocytic astrocytoma[33] and the most common malignant brain tumor in children, comprising 14.5% of newly diagnosed brain tumors.[34] In adults, medulloblastoma is rare, comprising fewer than 2% of CNS malignancies.[35]
The rate of new cases of childhood medulloblastoma is higher in males (62%) than females (38%), a feature that is not seen in adults.[32][36] Medulloblastoma and other PNET`s are more prevalent in younger children than older children. About 40% of medulloblastoma patients are diagnosed before the age of five, 31% are between the ages of 5 and 9, 18.3% are between the ages of 10 and 14, and 12.7% are between the ages of 15 and 19.[37]
Research models
Using gene transfer of SV40 large T-antigen in neuronal precursor cells of rats, a brain tumor model was established. The PNETs were histologically indistinguishable from the human counterparts and have been used to identify new genes involved in human brain tumor carcinogenesis.[38] The model was used to confirm p53 as one of the genes involved in human medulloblastomas, but since only about 10% of the human tumors showed mutations in that gene, the model can be used to identify the other binding partners of SV40 Large T- antigen, other than p53.[39] Recently a SHH-type mouse model with high incidence of medulloblastoma, a Patched 1 heterozygous mice knockout for the medulloblastoma suppressor Tis21 (Patched1+-/Tis21 KO).[40] The high medulloblastoma frequency appears to be caused by the down regulation of Cxcl3, being Cxcl3 induced by Tis21.[40] Consistently, the treatment with Cxcl3 completely prevents the growth of medulloblastoma lesions in a Shh-type mouse model of medulloblastoma.[41] Thus, CXCL3 is a target for medulloblastoma therapy.
References
- Roussel MF, Hatten ME (2011). Cerebellum development and medulloblastoma. Current Topics in Developmental Biology. Vol. 94. pp. 235–82. doi:10.1016/B978-0-12-380916-2.00008-5. ISBN 9780123809162. PMC 3213765. PMID 21295689.
- Hinz C, Hesser D (2006). Focusing On Brain Tumors: Medulloblastoma. American Brain Tumor Association. ISBN 0-944093-67-1. Archived from the original on 2008-09-08. Retrieved 2007-03-09.
- Smoll NR (March 2012). "Relative survival of childhood and adult medulloblastomas and primitive neuroectodermal tumors (PNETs)". Cancer. 118 (5): 1313–22. doi:10.1002/cncr.26387. PMID 21837678. S2CID 8490276.
- Polkinghorn WR, Tarbell NJ (May 2007). "Medulloblastoma: tumorigenesis, current clinical paradigm, and efforts to improve risk stratification". Nature Clinical Practice. Oncology. 4 (5): 295–304. doi:10.1038/ncponc0794. PMID 17464337. S2CID 24461280.
- Packer R (2002). "Medulloblastoma". Clinical Trials and Noteworthy Treatments for Brain Tumors.
- "Medulloblastoma". The Lecturio Medical Concept Library. Retrieved 10 August 2021.
- White LE, Levy RM, Alam M (2008). "Ch. 127. Neoplasias and Hyperplasias of Muscular and Neural Origin". In Wolff K, Goldsmith LA, Katz SI, Gilchrest B, Paller AS, Leffell DJ (eds.). Fitzpatrick's Dermatology in General Medicine (7e ed.). McGraw-Hill Medical.
- Ropper AH, Samuels MA. "Ch. 31. Intracranial Neoplasms and Paraneoplastic Disorders". In Ropper AH, Samuels MA (eds.). Adams and Victor's Principles of Neurology (9e ed.).
- Taylor MD, Northcott PA, Korshunov A, Remke M, Cho YJ, Clifford SC, et al. (April 2012). "Molecular subgroups of medulloblastoma: the current consensus". Acta Neuropathologica. 123 (4): 465–72. doi:10.1007/s00401-011-0922-z. PMC 3306779. PMID 22134537.
- Dubuc AM, Morrissy AS, Kloosterhof NK, Northcott PA, Yu EP, Shih D, et al. (April 2012). "Subgroup-specific alternative splicing in medulloblastoma". Acta Neuropathologica. 123 (4): 485–499. doi:10.1007/s00401-012-0959-7. PMC 3984840. PMID 22358458.
- Marino S (January 2005). "Medulloblastoma: developmental mechanisms out of control". Trends in Molecular Medicine. 11 (1): 17–22. doi:10.1016/j.molmed.2004.11.008. PMID 15649818.
- Gibson P, Tong Y, Robinson G, Thompson MC, Currle DS, Eden C, et al. (December 2010). "Subtypes of medulloblastoma have distinct developmental origins". Nature. 468 (7327): 1095–9. Bibcode:2010Natur.468.1095G. doi:10.1038/nature09587. PMC 3059767. PMID 21150899.
- Jones DT, Jäger N, Kool M, Zichner T, Hutter B, Sultan M, et al. (August 2012). "Dissecting the genomic complexity underlying medulloblastoma". Nature. 488 (7409): 100–5. Bibcode:2012Natur.488..100J. doi:10.1038/nature11284. PMC 3662966. PMID 22832583.
- Ellison DW (September 2010). "Childhood medulloblastoma: novel approaches to the classification of a heterogeneous disease". Acta Neuropathologica. 120 (3): 305–16. doi:10.1007/s00401-010-0726-6. PMID 20652577. S2CID 29093769.
- Cho YJ, Tsherniak A, Tamayo P, Santagata S, Ligon A, Greulich H, et al. (April 2011). "Integrative genomic analysis of medulloblastoma identifies a molecular subgroup that drives poor clinical outcome". Journal of Clinical Oncology. 29 (11): 1424–30. doi:10.1200/JCO.2010.28.5148. PMC 3082983. PMID 21098324.
- Northcott PA, Shih DJ, Peacock J, Garzia L, Morrissy AS, Zichner T, et al. (August 2012). "Subgroup-specific structural variation across 1,000 medulloblastoma genomes". Nature. 488 (7409): 49–56. Bibcode:2012Natur.488...49N. doi:10.1038/nature11327. PMC 3683624. PMID 22832581.
- Hatten ME, Roussel MF (March 2011). "Development and cancer of the cerebellum". Trends in Neurosciences. 34 (3): 134–42. doi:10.1016/j.tins.2011.01.002. PMC 3051031. PMID 21315459.
- Burger PC, Yu IT, Tihan T, Friedman HS, Strother DR, Kepner JL, et al. (September 1998). "Atypical teratoid/rhabdoid tumor of the central nervous system: a highly malignant tumor of infancy and childhood frequently mistaken for medulloblastoma: a Pediatric Oncology Group study". The American Journal of Surgical Pathology. 22 (9): 1083–92. doi:10.1097/00000478-199809000-00007. PMID 9737241.
- Merchant TE, Hua CH, Shukla H, Ying X, Nill S, Oelfke U (July 2008). "Proton versus photon radiotherapy for common pediatric brain tumors: comparison of models of dose characteristics and their relationship to cognitive function". Pediatric Blood & Cancer. 51 (1): 110–7. doi:10.1002/pbc.21530. PMID 18306274. S2CID 36735536.
- Blomstrand M, Brodin NP, Munck Af Rosenschöld P, Vogelius IR, Sánchez Merino G, Kiil-Berthlesen A, et al. (July 2012). "Estimated clinical benefit of protecting neurogenesis in the developing brain during radiation therapy for pediatric medulloblastoma". Neuro-Oncology. 14 (7): 882–9. doi:10.1093/neuonc/nos120. PMC 3379806. PMID 22611031.
- Eibl RH, Schneemann, M (Sep 2022). "Liquid biopsy for monitoring medulloblastoma". Extracell Vesicles Circ Nucleic Acids. 3: 263–74. doi:10.20517/evcna.2022.36.
- Michiels EM, Schouten-Van Meeteren AY, Doz F, Janssens GO, van Dalen EC (January 2015). "Chemotherapy for children with medulloblastoma". The Cochrane Database of Systematic Reviews. 1: CD006678. doi:10.1002/14651858.CD006678.pub2. PMID 25879092.
- Fossati P, Ricardi U, Orecchia R (February 2009). "Pediatric medulloblastoma: toxicity of current treatment and potential role of protontherapy". Cancer Treatment Reviews. 35 (1): 79–96. doi:10.1016/j.ctrv.2008.09.002. PMID 18976866.
- Crawford JR, MacDonald TJ, Packer RJ (December 2007). "Medulloblastoma in childhood: new biological advances". The Lancet. Neurology. 6 (12): 1073–85. doi:10.1016/S1474-4422(07)70289-2. PMID 18031705. S2CID 13013757.
- Pfister S, Remke M, Benner A, Mendrzyk F, Toedt G, Felsberg J, et al. (April 2009). "Outcome prediction in pediatric medulloblastoma based on DNA copy-number aberrations of chromosomes 6q and 17q and the MYC and MYCN loci". Journal of Clinical Oncology. 27 (10): 1627–36. doi:10.1200/JCO.2008.17.9432. PMID 19255330. S2CID 21794571.
- Packer, Roger J. (2010). "Medulloblastoma".
- "Identifying Low-Risk Medulloblastoma to De-escalate Therapy". Medscape. Retrieved 2020-01-03.
- Clinical trial number NCT02066220 for "International Society of Paediatric Oncology (SIOP) PNET 5 Medulloblastoma" at ClinicalTrials.gov
- Lannering B, Rutkowski S, Doz F, Pizer B, Gustafsson G, Navajas A, et al. (September 2012). "Hyperfractionated versus conventional radiotherapy followed by chemotherapy in standard-risk medulloblastoma: results from the randomized multicenter HIT-SIOP PNET 4 trial". Journal of Clinical Oncology. 30 (26): 3187–93. doi:10.1200/JCO.2011.39.8719. PMID 22851561.
- Sabel M, Fleischhack G, Tippelt S, Gustafsson G, Doz F, Kortmann R, et al. (September 2016). "Relapse patterns and outcome after relapse in standard risk medulloblastoma: a report from the HIT-SIOP-PNET4 study". Journal of Neuro-Oncology. 129 (3): 515–524. doi:10.1007/s11060-016-2202-1. PMC 5020107. PMID 27423645.
- Jakacki RI, Burger PC, Zhou T, Holmes EJ, Kocak M, Onar A, et al. (July 2012). "Outcome of children with metastatic medulloblastoma treated with carboplatin during craniospinal radiotherapy: a Children's Oncology Group Phase I/II study". Journal of Clinical Oncology. 30 (21): 2648–53. doi:10.1200/JCO.2011.40.2792. PMC 4559602. PMID 22665539.
- Smoll NR, Drummond KJ (November 2012). "The incidence of medulloblastomas and primitive neurectodermal tumours in adults and children". Journal of Clinical Neuroscience. 19 (11): 1541–4. doi:10.1016/j.jocn.2012.04.009. PMID 22981874. S2CID 7922631.
- "Chapter 7: Tumors of the Central Nervous System". Neuropathology. NEOMED. Archived from the original on 12 March 2012.
- Gurney JG, Smith MA, Bunin GR (1999). "CNS and Miscellaneous Intracranial and Intraspinal Neoplasms" (PDF). In Ries LA, Smith MA, Gurney JG, Linet M, Tamra T, Young JL, Bunin GR (eds.). Cancer Incidence and Survival among Children and Adolescents: United States SEER Program 1975–1995 (PDF). Bethesda MD: National Cancer Institute. NIH Pub. No. 99-4649.
- "Selected Primary Brain and Central Nervous System Tumor Age-Specific Incidence Rates" (PDF). Central Brain Tumor Registry of the United States, 1998–2002.
- "Selected Childhood Primary Brain and Central Nervous System Tumor Incidence Rates by Major Histology Groupings, Histology and Gender" (PDF). Central Brain Tumor Registry of the United States, 1998–2002.
- "Selected Childhood Primary Brain and Central Nervous System Tumor Age-Specific Incidence Rates" (PDF). Central Brain Tumor Registry of the United States, 1998–2002.
- Eibl RH, Kleihues P, Jat PS, Wiestler OD (March 1994). "A model for primitive neuroectodermal tumors in transgenic neural transplants harboring the SV40 large T antigen". The American Journal of Pathology. 144 (3): 556–64. PMC 1887088. PMID 8129041.
- Ohgaki H, Eibl RH, Wiestler OD, Yasargil MG, Newcomb EW, Kleihues P (November 1991). "p53 mutations in nonastrocytic human brain tumors". Cancer Research. 51 (22): 6202–5. PMID 1933879.
- Farioli-Vecchioli S, Cinà I, Ceccarelli M, Micheli L, Leonardi L, Ciotti MT, et al. (October 2012). "Tis21 knock-out enhances the frequency of medulloblastoma in Patched1 heterozygous mice by inhibiting the Cxcl3-dependent migration of cerebellar neurons". The Journal of Neuroscience. 32 (44): 15547–64. doi:10.1523/JNEUROSCI.0412-12.2012. PMC 6621585. PMID 23115191.
- Ceccarelli M, Micheli L, Tirone F (2016). "Suppression of Medulloblastoma Lesions by Forced Migration of Preneoplastic Precursor Cells with Intracerebellar Administration of the Chemokine Cxcl3". Frontiers in Pharmacology. 7: 484. doi:10.3389/fphar.2016.00484. PMC 5159413. PMID 28018222.