Cheyne–Stokes respiration
Cheyne–Stokes respiration is an abnormal pattern of breathing characterized by progressively deeper, and sometimes faster, breathing followed by a gradual decrease that results in a temporary stop in breathing called an apnea. The pattern repeats, with each cycle usually taking 30 seconds to 2 minutes.[1] It is an oscillation of ventilation between apnea and hyperpnea with a crescendo-diminuendo pattern, and is associated with changing serum partial pressures of oxygen and carbon dioxide.[2]
| Cheyne–Stokes respiration | |
|---|---|
 | |
| Graph showing Cheyne–Stokes respiration (bottom) and other pathological breathing patterns | |
| Pronunciation |
|
Cheyne–Stokes respiration and periodic breathing are the two regions on a spectrum of severity of oscillatory tidal volume. The distinction lies in what is observed at the trough of ventilation: Cheyne–Stokes respiration involves apnea (since apnea is a prominent feature in their original description) while periodic breathing involves hypopnea (abnormally small but not absent breaths).
These phenomena can occur during wakefulness or during sleep, where they are called the central sleep apnea syndrome (CSAS).[3]
It may be caused by damage to respiratory centers,[4] or by physiological abnormalities in congestive heart failure,[5] and is also seen in newborns with immature respiratory systems and in visitors new to high altitudes. One example is the breathing pattern in Joubert syndrome and related disorders.
Pathophysiology
Causes may include heart failure, kidney failure, narcotic poisoning, and raised intracranial pressure. The pathophysiology of Cheyne–Stokes breathing can be summarized as apnea leading to increased CO2 which causes excessive compensatory hyperventilation, in turn causing decreased CO2 which causes apnea, restarting the cycle.
In heart failure, the mechanism of the oscillation is unstable feedback in the respiratory control system. In normal respiratory control, negative feedback allows a steady level of alveolar gas concentrations to be maintained, and therefore stable tissue levels of oxygen and carbon dioxide (CO2). At the steady state, the rate of production of CO2 equals the net rate at which it is exhaled from the body, which (assuming no CO2 in the ambient air) is the product of the alveolar ventilation and the end-tidal CO2 concentration. Because of this interrelationship, the set of possible steady states forms a hyperbola:
- Alveolar ventilation = body CO2 production/end-tidal CO2 fraction.
In the figure below, this relationship is the curve falling from the top left to the bottom right. Only positions along this curve permit the body's CO2 production to be exactly compensated for by exhalation of CO2. Meanwhile, there is another curve, shown in the figure for simplicity as a straight line from bottom left to top right, which is the body's ventilatory response to different levels of CO2. Where the curves cross is the potential steady state (S).
Through respiratory control reflexes, any small transient fall in ventilation (A) leads to a corresponding small rise (A') in alveolar CO2 concentration which is sensed by the respiratory control system so that there is a subsequent small compensatory rise in ventilation (B) above its steady state level (S) that helps restore CO2 back to its steady state value. In general, transient or persistent disturbances in ventilation, CO2 or oxygen levels can be counteracted by the respiratory control system in this way.
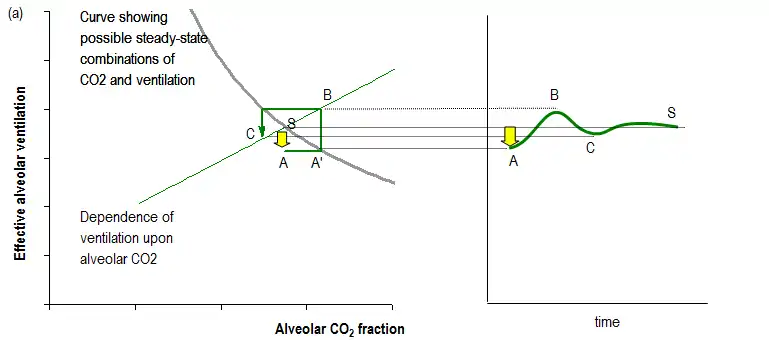
However, in some pathological states, the feedback is more powerful than is necessary to simply return the system towards its steady state. Instead, ventilation overshoots and can generate an opposite disturbance to the original disturbance. If this secondary disturbance is larger than the original, the next response will be even larger, and so on, until very large oscillations have developed, as shown in the figure below.
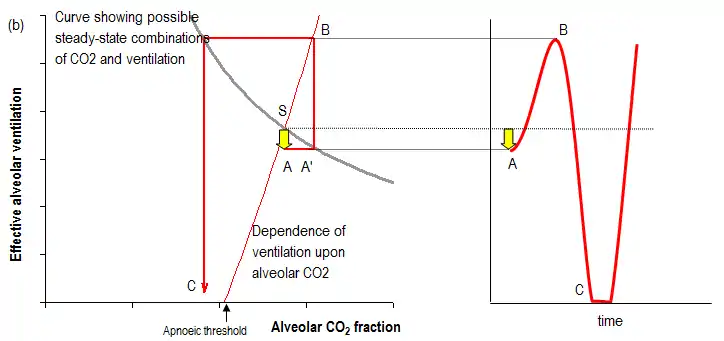
The cycle of enlargement of disturbances reaches a limit when successive disturbances are no longer larger, which occurs when physiological responses no longer increase linearly in relation to the size of the stimulus. The most obvious example of this is when ventilation falls to zero: it cannot be any lower. Thus Cheyne–Stokes respiration can be maintained over periods of many minutes or hours with a repetitive pattern of apneas and hyperpneas.
The end of the linear decrease in ventilation in response to falls in CO2 is not, however, at apnea. It occurs when ventilation is so small that air being breathed in never reaches the alveolar space, because the inspired tidal volume is no larger than the volume of the large airways such as the trachea. Consequently, at the nadir of periodic breathing, ventilation of the alveolar space may be effectively zero; the easily observable counterpart of this is failure at that time point of the end-tidal gas concentrations to resemble realistic alveolar concentrations.
Many potential contributory factors have been identified by clinical observation, but unfortunately they are all interlinked and co-vary extensively. Widely accepted risk factors are hyperventilation, prolonged circulation time, and reduced blood gas buffering capacity.[6][7]
They are physiologically interlinked in that (for any given patient) circulation time decreases as cardiac output increases. Likewise, for any given total body CO2 production rate, alveolar ventilation is inversely proportional to end-tidal CO2 concentration (since their mutual product must equal total body CO2 production rate). Chemoreflex sensitivity is closely linked to the position of the steady state, because if chemoreflex sensitivity increases (other things being equal) the steady-state ventilation will rise and the steady-state CO2 will fall. Because ventilation and CO2 are easy to observe, and because they are commonly measured clinical variables which do not require any particular experiment to be conducted in order to observe them, abnormalities in these variables are more likely to be reported in the literature. However, other variables, such as chemoreflex sensitivity can only be measured by specific experiment, and therefore abnormalities in them will not be found in routine clinical data.[8] When measured in patients with Cheyne–Stokes respiration, hypercapnic ventilatory responsiveness may be elevated by 100% or more. When not measured, its consequences—such as a low mean PaCO2 and elevated mean ventilation—may sometimes appear to be the most prominent feature.[9]
Associated conditions
This abnormal pattern of breathing, in which breathing is absent for a period and then rapid for a period, can be seen in patients with heart failure,[10][11] strokes, hyponatremia, traumatic brain injuries, and brain tumors. In some instances, it can occur in otherwise healthy people during sleep at high altitudes. It can occur in all forms of toxic metabolic encephalopathy.[12] It is a symptom of carbon monoxide poisoning, along with syncope or coma. This type of respiration is different from respiratory depression, often seen after morphine administration.[13]
Hospices sometimes document the presence of Cheyne–Stokes breathing as a patient nears death, and report that patients able to speak after such episodes do not report any distress associated with the breathing, although it is sometimes disturbing to the family.
Related patterns
Cheyne–Stokes respirations are not the same as Biot's respirations ("cluster breathing"), in which groups of breaths tend to be similar in size.
They differ from Kussmaul respirations in that the Kussmaul pattern is one of consistent very deep breathing at a normal or increased rate.
History
The condition was named after John Cheyne and William Stokes, the physicians who first described it in the 19th century.[14][15]
The term became widely known and used in the Soviet Union after the death of Joseph Stalin in 1953, because the Soviet press announced that the ailing Stalin had Cheyne–Stokes respiration.[16]
References
- "Cheynes–Stokes Respiration". WebMD LLC. Retrieved 2010-10-05.
- "Cheyne–Stokes respiration". WrongDiagnosis.com. Health Grades Inc. Retrieved 2010-09-03.
- Kumar, Parveen; Clark, Michael (2005). "13". Clinical Medicine (6 ed.). Elsevier Saunders. p. 733. ISBN 978-0-7020-2763-5.
- "Cheyne-Stokes respiration" at Dorland's Medical Dictionary
- Francis, DP; Willson, K; Davies, LC; Coats, AJ; Piepoli, M (2000). "Quantitative general theory for periodic breathing in heart failure and its clinical implications" (PDF). Circulation. 102 (18): 2214–2221. CiteSeerX 10.1.1.505.7194. doi:10.1161/01.cir.102.18.2214. PMID 11056095. Retrieved 2010-09-05.
- Khoo, MC; Gottschalk, A; Pack, AI (1991). "Sleep-induced periodic breathing and apnea: a theoretical study". Journal of Applied Physiology. 70 (5): 2014–24. doi:10.1152/jappl.1991.70.5.2014. PMID 1907602.
- Naughton, M. T. (1998). "Pathophysiology and treatment of Cheyne-Stokes respiration". Thorax. 53 (6): 514–518. doi:10.1136/thx.53.6.514. PMC 1745239. PMID 9713454.
- Manisty CH, Willson K, Wensel R, Whinnett ZI, Davies JE, Oldfield WL, Mayet J, Francis DP (2006). "Development of respiratory control instability in heart failure: a novel approach to dissect the pathophysiological mechanisms". J. Physiol. 577 (Pt 1): 387–401. doi:10.1113/jphysiol.2006.116764. PMC 1804209. PMID 16959858.
- Wilcox, I.; Grunstein, R. R.; Collins, F. L.; Berthon-Jones, M.; Kelly, D. T.; Sullivan, C. E. (1993). "The role of central chemosensitivity in central apnea of heart failure". Sleep. 16 (8 Suppl): S37–S38. doi:10.1093/sleep/16.suppl_8.S37. PMID 8178021.
- Lanfranchi PA, Braghiroli A, Bosimini E, et al. (March 1999). "Prognostic value of nocturnal Cheyne–Stokes respiration in chronic heart failure". Circulation. 99 (11): 1435–40. doi:10.1161/01.cir.99.11.1435. PMID 10086966.
- Brack T, Thüer I, Clarenbach CF, et al. (November 2007). "Daytime Cheyne–Stokes respiration in ambulatory patients with severe congestive heart failure is associated with increased mortality". Chest. 132 (5): 1463–71. doi:10.1378/chest.07-0121. PMID 17646230.
- The Diagnosis of Stupor and Coma by Plum and Posner, ISBN 0-19-513898-8
- Davis, Mellar (June 30, 2018). "Opioids, Dyspnea and Risks" (PDF). mascc.org. Retrieved December 17, 2020.
- J. Cheyne: A case of apoplexy in which the fleshy part of the heart was converted into fat. Dublin Hospital Reports, 1818, 2: 216-223. Reprinted in F. A. Willius & T. E. Keys: Cardiac Classics, 1941, pp. 317-320
- William Stokes (1854), "Fatty degeneration of the heart." In his: The Diseases of the Heart and Aorta Dublin, by Alok Mishra pp. 320–327.
- Gessen, Masha (6 March 2018). ""The Death of Stalin" Captures the Terrifying Absurdity of a Tyrant". The New Yorker.