Congenital pulmonary airway malformation
Congenital pulmonary airway malformation (CPAM), formerly known as congenital cystic adenomatoid malformation (CCAM), is a congenital disorder of the lung similar to bronchopulmonary sequestration. In CPAM, usually an entire lobe of lung is replaced by a non-working cystic piece of abnormal lung tissue. This abnormal tissue will never function as normal lung tissue. The underlying cause for CPAM is unknown. It occurs in approximately 1 in every 30,000 pregnancies.[1]
| Congenital pulmonary airway malformation | |
|---|---|
 | |
| Specialty | Medical genetics |
In most cases the outcome of a fetus with CPAM is very good. In rare cases, the cystic mass grows so large as to limit the growth of the surrounding lung and cause pressure against the heart. In these situations, the CPAM can be life-threatening for the fetus. CPAM can be separated into five types, based on clinical and pathologic features.[2] CPAM type 1 is the most common, with large cysts and a good prognosis. CPAM type 2 (with medium-sized cysts) often has a poor prognosis, owing to its frequent association with other significant anomalies. Other types are rare.[3]
Signs and symptoms
Three quarters of affected patients are asymptomatic. However, 25% develop cyanosis, pneumothorax, and show signs of increased breathing difficulty (tachypnoea and intercostal retractions).At examination, they may show hyper-resonance at percussion, diminished vesicular murmur and an asymmetrical thorax.
Cause
The cause of CPAM is unknown.
Diagnosis

CPAMs are often identified during routine prenatal ultrasonography. Identifying characteristics on the sonogram include: an echogenic (bright) mass appearing in the chest of the fetus, displacement of the heart from its normal position, a flat or everted (pushed downward) diaphragm, or the absence of visible lung tissue.
CPAMs are classified into three different types based largely on their gross appearance. Type I has a large (>2 cm) multiloculated cysts. Type II has smaller uniform cysts. Type III is not grossly cystic, referred to as the "adenomatoid" type. Microscopically, the lesions are not true cysts, but communicate with the surrounding parenchyma. Some lesions have an abnormal connection to a blood vessel from an aorta and are referred to as "hybrid lesions."
Imaging
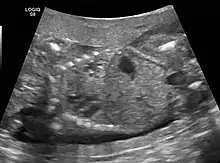

The earliest point at which a CPAM can be detected is by prenatal ultrasound. The classic description is of an echogenic lung mass that gradually disappears over subsequent ultrasounds. The disappearance is due to the malformation becoming filled with fluid over the course of the gestation, allowing the ultrasound waves to penetrate it more easily and rendering it invisible on sonographic imaging. When a CPAM is rapidly growing, either solid or with a dominant cyst, they have a higher incidence of developing venous outflow obstruction, cardiac failure and ultimately hydrops fetalis. If hydrops is not present, the fetus has a 95% chance of survival. When hydrops is present, risk of fetal demise is much greater without in utero surgery to correct the pathophysiology. The greatest period of growth is during the end of the second trimester, between 20–26 weeks.
A measure of mass volume divided by head circumference, termed cystic adenomatoid malformation volume ratio (CVR) has been developed to predict the risk of hydrops. The lung mass volume is determined using the formula (length × width × anteroposterior diameter ÷ 2), divided by head circumference. With a CVR greater than 1.6 being considered high risk. Fetuses with a CVR less than 1.6 and without a dominant cyst have less than a 3% risk of hydrops. After delivery, if the patient is symptomatic, resection is mandated. If the infant is asymptomatic, the need for resection is a subject of debate, though it is usually recommended. Development of recurrent infections, rhabdomyosarcoma, adenocarcinomas in situ within the lung malformation have been reported.[4]
Treatment
In most cases, a fetus with CPAM is closely monitored during pregnancy and the CPAM is removed via surgery after birth.[5] Most babies with a CPAM are born without complication and are monitored during the first few months. Many patients have surgery, typically before their first birthday, because of the risk of recurrent lung infections associated with CPAMs. Some pediatric surgeons can safely remove these lesions using very tiny incisions using minimally invasive surgical techniques (thoracoscopy). However, some CPAM patients live a full life without any complication or incident. It is hypothesized that there are thousands of people living with an undetected CPAM. Through ultrasound testing employed in recent years, many more patients are aware that they live with this condition. Rarely, long standing CPAMs have been reported to become cancerous.
Very large cystic masses might pose a danger during birth because of the airway compression. In this situation, a special surgical type of delivery called the EXIT procedure may be used.
In rare extreme cases, where fetus's heart is in danger, fetal surgery can be performed to remove the CPAM. If non-immune hydrops fetalis develop, there is a near universal mortality of the fetus without intervention. Fetal surgery can improve the chances of survival to 50-60%. Recently, several studies found that a single course of prenatal steroids (betamethasone) may increase survival in hydropic fetuses with microcystic CPAMs to 75-100%.[6][7] These studies indicate that large microcystic lesions may be treated prenatally without surgical intervention. Large macrocyst lesions may require in utero placement of a Harrison thoracoamniotic shunt.
References
- http://synapse.koreamed.org/Synapse/Data/PDFData/0003TRD/trd-72-501.pdf
- Nihal Kilinç et al., "Congenital Pulmonary Airway Malformation: Case Report". Perinatal Journal, vol. 15, issue 1 (April 2007)
- Robbins and Cotran, Pathologic Basis of Disease 7th ed.
- Coley, editor-in-chief Brian D. (2013). Caffey's pediatric diagnostic imaging (Twelfth ed.). Philadelphia: Elsevier Saunders. ISBN 978-0323081764.
{{cite book}}:|first1=has generic name (help) - Ehrenberg-Buchner, S; Stapf, AM; Berman, DR; Drongowski, RA; Mychaliska, GB; Treadwell, MC; Kunisaki, SM (Nov 15, 2012). "Fetal lung lesions: can we start to breathe easier?". American Journal of Obstetrics and Gynecology. 208 (2): 151.e1–7. doi:10.1016/j.ajog.2012.11.012. PMID 23159697.
- Curran PF, Jelin EB, Rand L, Hirose S, Feldstein VA, Goldstein RB, Lee H (2010). "Prenatal steroids for microcystic congenital cystic adenomatoid malformations". J Pediatr Surg. 45 (1): 145–50. doi:10.1016/j.jpedsurg.2009.10.025. PMID 20105595.
- Peranteau WH, Wilson RD, Liechty KW, Johnson MP, Bebbington MW, Hedrick HL, Flake AW, Adzick NS (2007). "Effect of maternal betamethasone administration on prenatal congenital cystic adenomatoid malformation growth and fetal survival". Fetal Diagn Ther. 22 (5): 365–371. doi:10.1159/000103298. PMID 17556826.