Focal segmental glomerulosclerosis
Focal segmental glomerulosclerosis (FSGS) is a histopathologic finding of scarring (sclerosis) of glomeruli and damage to renal podocytes.[2][3] This process damages the filtration function of the kidney, resulting in protein loss in the urine.[3] FSGS is a leading cause of excess protein loss—nephrotic syndrome—in children and adults.[4] Signs and symptoms include proteinuria, water retention, and edema.[2][5] Kidney failure is a common long-term complication of disease.[5][6] FSGS can be classified as primary versus secondary depending on whether a particular toxic or pathologic stressor can be identified as the cause.[7][8] Diagnosis is established by renal biopsy,[2][9] and treatment consists of glucocorticoids and other immune-modulatory drugs.[10] Response to therapy is variable, with a significant portion of patients progressing to end-stage kidney failure.[5] FSGS is estimated to occur in 2-3 persons per million, with males and African peoples at higher risk .[11][12]
| Focal segmental glomerulosclerosis | |
|---|---|
| Other names | focal glomerular sclerosis,[1] focal nodular glomerulosclerosis[1] |
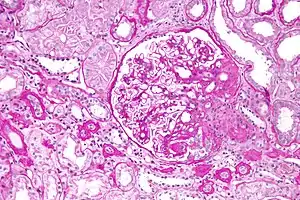 | |
| Light micrograph of focal segmental glomerulosclerosis, hilar variant. Kidney biopsy. PAS stain. | |
| Specialty | Nephrology |
Signs and symptoms
The most common symptoms are a result of abnormal loss of protein from the glomerulus of the kidney, and include:[2][5]
- Frothy urine (due to excess protein)
- Excess water retention (pitting edema, due to loss of serum albumin)
- Susceptibility to infection (due to loss of serum antibodies)
Common signs are also due to loss of blood proteins by the glomerulus of the kidney, including:[2][5][9]
- Protein in the urine (often in the nephrotic syndrome-range of >3.5 g/day)
- Low serum albumin (<3.5 g/dl)
- Low serum antibodies
- High serum cholesterol (compensatory by the liver to compensate for low serum oncotic pressure)
- Fatty casts in the urine (secondary to hypercholesterolemia)
Pathophysiology

FSGS is primarily a disease of the renal glomerulus, the site of filtration of ions and solutes.[13][14] Podocytes are specialized cells lining the Bowman's capsule that contribute to the filtration barrier, preventing molecules larger than 5 nm from being filtered.[15] FSGS involves damage to the renal podocytes such that larger molecules, most notably proteins, are filtered and lost through the kidney.[16][17] Thus, many of the signs and symptoms of FSGS are related to protein loss.[18]
On histology, FSGS manifests as damage (sclerosis) to segments of glomeruli; moreover, only a portion of glomeruli are affected.[19][20] The focal and segmental nature of disease seen on histology help to distinguish FSGS from other types of glomerular sclerosis.[20]
FSGS can be classified by the putative cause of damage to podocytes. Primary FSGS involves cases in which no cause is readily identifiable.[21] It is presumed that a set of unidentified circulating factors in the blood contribute to podocyte damage in these cases.[21][22]
Secondary FSGS is caused by an identifiable stress or toxin that injures podocytes.[21] Many causes of secondary FSGS contribute to podocyte injury through hyperfiltration, which is a scenario of excess filtration by renal glomeruli.[23] Hyperfiltration can be caused by obesity or loss of the contralateral kidney, among other causes.[23]
Secondary FSGS can also be caused by toxins, including steroids and heroin.[24][25]
A number of genes have been implicated in FSGS. These include: NPHS1, which encodes the protein nephrin that contributes to the filtration barrier;[26] NPHS2, which encodes the protein podocin found in podocytes;[27] and INF2, which encodes the actin-binding protein formin.[28]
The pathogenesis of HIV-associated FSGS is unclear, but may be primarily due to the presence of the G1/G2 risk alleles for APOL1. There is some data to suggest that HIV can infect tubular epithelial cells and podocytes, but much remains to be known.[29]
Diagnosis
Diagnosis of FSGS is made by renal biopsy that includes at least 15 serial cuts with at least 8 glomeruli.[30][31] Histologic features include sclerosis (scarring) of a portion (average: 15%) of the glomerular space, with only a portion of glomeruli manifesting any sclerosis.[31]
Other tests helpful in the diagnosis include urine protein, urinalysis, serum albumin, and serum lipids.[2] A clinical picture of proteinuria, low blood protein levels (albumin, antibodies), and high blood cholesterol would support a diagnosis of FSGS, although these do not help to distinguish between FSGS and other causes of proteinuria.[5][9]
Classification
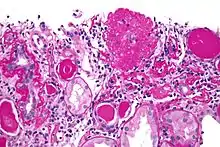
Five mutually exclusive variants of focal segmental glomerulosclerosis may be distinguished by the pathologic findings seen on renal biopsy:[32]
- Collapsing variant
- Glomerular tip lesion variant
- Cellular variant
- Perihilar variant
- Not otherwise specified (NOS) variant.
Recognition of these variants may have prognostic value in individuals with primary focal segmental glomerulosclerosis. The collapsing variant is associated with higher rate of progression to end-stage renal disease, whereas the glomerular tip lesion variant has a low rate of progression to end-stage renal disease in most patients.[8] The cellular variant shows similar clinical presentation to collapsing and glomerular tip variant but has intermediate outcomes between the other two variants.[8]
Treatment
First-line treatment for primary FSGS consists of anti-inflammatory drugs.[10] Specifically, glucocorticoids are begun in patients manifesting with nephrotic-range proteinuria (>3.5 g/day).[33][34] For patients who maintain nephrotic-range proteinuria despite glucocorticoids, or for patients who demonstrate glucocorticoid intolerance, calcineurin inhibitors (e.g., tacrolimus) are initiated.[34] Successful treatment is defined as a drop in proteinuria to sub-nephrotic ranges.[6]
The treatment of secondary FSGS involves addressing the particular toxic or stress agent.[33]
Prognosis
The majority of untreated cases of FSGS will progress to end-stage kidney disease.[35] Important prognostic factors include the degree of proteinuria and initial response to therapy.
Patients with nephrotic-range (>3.5 g/day) proteinuria have over a 50% rate of progression to end-stage kidney disease at 10 years.[6] Only 15% of patients with sub-nephrotic ranges of proteinuria progress to end-stage renal failure at 10 years.[6]
Initial response to therapy also dictates long-term outcomes. Those defined as having a "complete response" typically manifest a proteinuria of <300 mg/day; those with a "partial response" manifest a sub-nephrotic range of proteinuria, <3.5 g/day.[36] Either complete or partial response is associated with 80% kidney survival at 10-years, compared with about 50% among non-responsive patients.[36]
Epidemiology
FSGS accounts for 35% of all cases of nephrotic syndrome, making it one of the most common causes of nephrotic syndrome in the United States.[7] FSGS accounts for 2% of all cases of kidney failure.[4] African American patients have 4 times the likelihood of developing FSGS. Men are about 2 times as likely to develop FSGS as compared with women.[11]
See also
References
- "focal segmental glomerulosclerosis" at Dorland's Medical Dictionary.
- Rosenberg, Avi Z.; Kopp, Jeffrey B. (2017-03-07). "Focal Segmental Glomerulosclerosis". Clinical Journal of the American Society of Nephrology. 12 (3): 502–517. doi:10.2215/CJN.05960616. ISSN 1555-9041. PMC 5338705. PMID 28242845.
- D'Agati V. The many masks of focal segmental glomerulosclerosis. Kidney Int. 1994 Oct;46(4):1223-41. doi: 10.1038/ki.1994.388. PMID 7861720.
- Kitiyakara C, Eggers P, Kopp JB. Twenty-one-year trend in ESRD due to focal segmental glomerulosclerosis in the United States. Am J Kidney Dis. 2004 Nov;44(5):815-25. PMID 15492947.
- Rydel JJ, Korbet SM, Borok RZ, Schwartz MM. Focal segmental glomerular sclerosis in adults: presentation, course, and response to treatment. Am J Kidney Dis. 1995 Apr;25(4):534-42. doi: 10.1016/0272-6386(95)90120-5. PMID 7702047.
- Korbet SM, Schwartz MM, Lewis EJ. Primary focal segmental glomerulosclerosis: clinical course and response to therapy. Am J Kidney Dis. 1994 Jun;23(6):773-83. doi: 10.1016/s0272-6386(12)80128-4. PMID 8203357.
- Haas M, Meehan SM, Karrison TG, Spargo BH. Changing etiologies of unexplained adult nephrotic syndrome: a comparison of renal biopsy findings from 1976-1979 and 1995-1997. Am J Kidney Dis. 1997 Nov;30(5):621-31. doi: 10.1016/s0272-6386(97)90485-6. PMID 9370176.
- Fogo AB. Causes and pathogenesis of focal segmental glomerulosclerosis. Nat Rev Nephrol. 2015 Feb;11(2):76-87. doi: 10.1038/nrneph.2014.216. Epub 2014 Dec 2. PMID 25447132; PMCID: PMC4772430.
- Kiffel J, Rahimzada Y, Trachtman H. Focal segmental glomerulosclerosis and chronic kidney disease in pediatric patients. Adv Chronic Kidney Dis. 2011 Sep;18(5):332-8. doi: 10.1053/j.ackd.2011.03.005. PMID 21896374; PMCID: PMC3709971.
- Campbell KN, Tumlin JA. Protecting Podocytes: A Key Target for Therapy of Focal Segmental Glomerulosclerosis. Am J Nephrol. 2018;47 Suppl 1(Suppl 1):14-29. doi: 10.1159/000481634. Epub 2018 May 31. PMID 29852493; PMCID: PMC6589822.
- Tucker JK. Focal segmental glomerulosclerosis in African Americans. Am J Med Sci. 2002 Feb;323(2):90-3. doi: 10.1097/00000441-200202000-00006. PMID 11863085.
- Kitiyakara C, Kopp JB, Eggers P. Trends in the epidemiology of focal segmental glomerulosclerosis. Semin Nephrol. 2003 Mar;23(2):172-82. doi: 10.1053/snep.2003.50025. PMID 12704577.
- Wallace MA. Anatomy and physiology of the kidney. AORN J. 1998 Nov;68(5):800, 803-16, 819-20; quiz 821-4. doi: 10.1016/s0001-2092(06)62377-6. PMID 9829131.
- Pollak MR, Quaggin SE, Hoenig MP, Dworkin LD. The glomerulus: the sphere of influence. Clin J Am Soc Nephrol. 2014 Aug 7;9(8):1461-9. doi: 10.2215/CJN.09400913. Epub 2014 May 29. PMID 24875196; PMCID: PMC4123398.
- Tojo A, Kinugasa S. Mechanisms of glomerular albumin filtration and tubular reabsorption. Int J Nephrol. 2012;2012:481520. doi: 10.1155/2012/481520. Epub 2012 May 20. PMID 22685655; PMCID: PMC3363986.
- Nagata M. Podocyte injury and its consequences. Kidney Int. 2016 Jun;89(6):1221-30. doi: 10.1016/j.kint.2016.01.012. Epub 2016 Mar 19. PMID 27165817.
- Wang CS, Greenbaum LA. Nephrotic Syndrome. Pediatr Clin North Am. 2019 Feb;66(1):73-85. doi: 10.1016/j.pcl.2018.08.006. PMID 30454752.
- "Focal Segmental Glomerulosclerosis (FSGS)". Cleveland Clinic. Retrieved 2022-06-30.
- Ichikawa I, Fogo A. Focal segmental glomerulosclerosis. Pediatr Nephrol. 1996 Jun;10(3):374-91. doi: 10.1007/BF00866790. PMID 8792409.
- Nagata M, Kobayashi N, Hara S. Focal segmental glomerulosclerosis; why does it occur segmentally? Pflugers Arch. 2017 Aug;469(7-8):983-988. doi: 10.1007/s00424-017-2023-x. Epub 2017 Jun 29. PMID 28664408.
- De Vriese AS, Sethi S, Nath KA, et al. Differentiating Primary, Genetic, and Secondary FSGS in Adults: A Clinicopathologic Approach. J Am Soc Nephrol 2018; 29:759.
- Rennke HG, Klein PS. Pathogenesis and significance of nonprimary focal and segmental glomerulosclerosis. Am J Kidney Dis 1989; 13:443.
- Helal I, Fick-Brosnahan GM, Reed-Gitomer B, Schrier RW. Glomerular hyperfiltration: definitions, mechanisms and clinical implications. Nat Rev Nephrol. 2012 Feb 21;8(5):293-300. doi: 10.1038/nrneph.2012.19. PMID 22349487.
- Dubrow A, Mittman N, Ghali V, Flamenbaum W. The changing spectrum of heroin-associated nephropathy. Am J Kidney Dis 1985; 5:36.
- Kasiske BL, Crosson JT. Renal disease in patients with massive obesity. Arch Intern Med 1986; 146:1105.
- Philippe A, Nevo F, Esquivel EL, Reklaityte D, Gribouval O, Tête MJ, Loirat C, Dantal J, Fischbach M, Pouteil-Noble C, Decramer S, Hoehne M, Benzing T, Charbit M, Niaudet P, Antignac C. Nephrin mutations can cause childhood-onset steroid-resistant nephrotic syndrome. J Am Soc Nephrol. 2008 Oct;19(10):1871-8. doi: 10.1681/ASN.2008010059. Epub 2008 Jul 9. PMID 18614772; PMCID: PMC2551572.
- Boute N, Gribouval O, Roselli S, Benessy F, Lee H, Fuchshuber A, Dahan K, Gubler MC, Niaudet P, Antignac C. NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat Genet. 2000 Apr;24(4):349-54. doi: 10.1038/74166. Erratum in: Nat Genet 2000 May;25(1):125. PMID 10742096.
- Brown EJ, Schlöndorff JS, Becker DJ, Tsukaguchi H, Tonna SJ, Uscinski AL, Higgs HN, Henderson JM, Pollak MR. Mutations in the formin gene INF2 cause focal segmental glomerulosclerosis. Nat Genet. 2010 Jan;42(1):72-6. doi: 10.1038/ng.505. Epub 2009 Dec 20. Erratum in: Nat Genet. 2010 Apr;42(4):361. Tonna, Stephen J [added]. PMID 20023659; PMCID: PMC2980844.
- Chang, Anthony, Robbins & Cotran Pathologic Basis of Disease, Chapter 20, 895-952
- Fuiano G, Comi N, Magri P, et al. Serial morphometric analysis of sclerotic lesions in primary "focal" segmental glomerulosclerosis. J Am Soc Nephrol 1996; 7:49.
- Schwartz MM, Korbet SM. Primary focal segmental glomerulosclerosis: pathology, histological variants, and pathogenesis. Am J Kidney Dis 1993; 22:874.
- Thomas DB, Franceschini N, Hogan SL, et al. (2006). "Clinical and pathologic characteristics of focal segmental glomerulosclerosis pathologic variants". Kidney Int. 69 (5): 920–6. doi:10.1038/sj.ki.5000160. PMID 16518352.
- Chen YM, Liapis H. Focal segmental glomerulosclerosis: molecular genetics and targeted therapies. BMC Nephrol. 2015 Jul 9;16:101. doi: 10.1186/s12882-015-0090-9. PMID 26156092; PMCID: PMC4496884.
- Raina R, Wang J, Sharma A, Chakraborty R. Extracorporeal Therapies in the Treatment of Focal Segmental Glomerulosclerosis. Blood Purif. 2020;49(5):513-523. doi: 10.1159/000506277. Epub 2020 Feb 19. PMID 32074606.
- Deegens JK, Assmann KJ, Steenbergen EJ, Hilbrands LB, Gerlag PG, Jansen JL, Wetzels JF. Idiopathic focal segmental glomerulosclerosis: a favourable prognosis in untreated patients? Neth J Med. 2005 Nov;63(10):393-8. PMID 16301760.
- Troyanov S, Wall CA, Miller JA, Scholey JW, Cattran DC; Toronto Glomerulonephritis Registry Group. Focal and segmental glomerulosclerosis: definition and relevance of a partial remission. J Am Soc Nephrol. 2005 Apr;16(4):1061-8. doi: 10.1681/ASN.2004070593. Epub 2005 Feb 16. PMID 15716334.