Hereditary diffuse leukoencephalopathy with spheroids
Hereditary diffuse leukoencephalopathy with spheroids (HDLS) is a rare adult onset autosomal dominant disorder characterized by cerebral white matter degeneration with demyelination and axonal spheroids leading to progressive cognitive and motor dysfunction. Spheroids are axonal swellings with discontinuous or absence of myelin sheaths. It is believed that the disease arises from primary microglial dysfunction that leads to secondary disruption of axonal integrity, neuroaxonal damage, and focal axonal spheroids leading to demyelination. Spheroids in HDLS resemble to some extent those produced by shear stress in a closed head injury with damage to axons, causing them to swell due to blockage of axoplasmic transport. In addition to trauma, axonal spheroids can be found in aged brain, stroke, and in other degenerative diseases.[1] In HDLS, it is uncertain whether demyelination occurs prior to the axonal spheroids or what triggers neurodegeneration after apparently normal brain and white matter development, although genetic deficits suggest that demyelination and axonal pathology may be secondary to microglial dysfunction.[2] The clinical syndrome in patients with HDLS is not specific and it can be mistaken for Alzheimer's disease, frontotemporal dementia, atypical Parkinsonism, multiple sclerosis, or corticobasal degeneration.[3]
| Hereditary diffuse leukoencephalopathy with spheroids (HDLS) | |
|---|---|
| Other names | Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia, Autosomal dominant leukoencephalopathy with neuroaxonal spheroids |
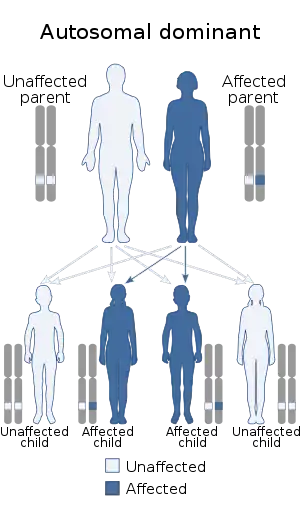 | |
| Hereditary diffuse leukoencephalopathy with spheroids is inherited in an autosomal dominant manner | |
Symptoms
With symptoms of personality changes, behavioral changes, dementia, depression, and epilepsy, HDLS has been commonly misdiagnosed for a number of other diseases.[4] Dementia or frontotemporal behavioral changes, for example, have commonly steered some clinicians to mistakenly consider diagnoses such as Alzheimer's disease, frontotemporal dementia or atypical Parkinsonism. The presence of white matter changes has led to misdiagnosis of multiple sclerosis. HDLS commonly manifests with neuropsychiatric symptoms, progressing to dementia, and after a few years shows motor dysfunction. Eventually patients become reliant on wheelchairs.[3]
White matter degeneration is associated with and makes differential diagnoses out of other adult onset leukodystrophies such as metachromatic leukodystrophy (MLD), Krabbe disease (globoid cell leukodystrophy), and X-linked adrenoleukodystrophy (X-ADL).[2]
| Disease | Exclusive Trait |
|---|---|
| MLD | Accumulation of metachromatic material in white matter |
| Krabbe Disease | Presence of globoid cells derived from microglia which have multiple nuclei |
| X-ALD | Predominant parieto-occipital white matter abnormality |
| Vanishing White Matter (VWM) Disease |
|
| Nasu-Hakola |
|
Neuropsychiatric symptoms
Many neuropsychiatric symptoms have been identified in clinical studies of HDLS patients. These include severe depression and anxiety that have been identified in about 70% of HDLS families, verging on suicidal tendencies and substance abuse such as alcoholism. Additionally, patients may exhibit disorientation, confusion, agitation, irritability, aggressiveness, an altered mental state, the loss of the ability to execute learned movements (apraxia), or the inability to speak (mutism).[3]
Motor impairment
Persons with HDLS can develop tremors, decreased body movement, unsteadiness (Parkinsonism, muscles on one side of the body in constant contraction (spastic hemiparesis), impairment in motor and sensory function in the lower extremities (paraparesis), paralysis resulting in partial or total loss of all extremities and torso (tetraparesis), and the lack of voluntary coordination of muscle movements (ataxia).[3]
Causes
The cause of HDLS in most families is mutation in the colony stimulating factor 1 receptor (CSF1R), a growth factor for microglia and monocyte/macrophages, suggesting that microglial dysfunction may be primary in HDLS.[4]
The mutations are concentrated in tyrosine kinase domain (TKD) of the protein. Mutations were mainly found in exons 12-22 of the intracellular TKD, including 10 missense mutations that have a single nucleotide deletion and a single codon deletion that consists of a triplet of nucleotides that have been removed causing a whole amino acid to not be coded. Additionally, three splice site mutations were identified that caused an in-frame deletion of an exon, an expressed nucleotide sequence, leading to the removal of more than 40 amino acids in the TKD.[4]
This determination has based upon genetic studies of 14 HDLS families confirming mutations in this gene. The CSF1 receptor protein primarily functions in regulation, survival, proliferation, and differentiation of microglial cells.[5] The mechanism of microglial dysfunction due to mutations in CSF1R to the myelin loss and axonal spheroid formation remains unknown. Further research is needed to better understand disease pathogenesis.
Pathology
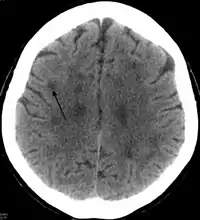
In HDLS, there is enlargement of the lateral ventricles and marked thinning or weakening of cerebral white matter.[6] The loss of white matter is caused by myelin loss. These changes are associated with diffuse gliosis, moderate loss of axons and many axonal spheroids.[1]
Activated or ameboid microglia and macrophages that contain myelin debris, lipid droplets and brown autofluorescent pigment granules are found in the areas with demyelination and axonal spheroids. In severely degenerated areas there are many large, reactive astrocytes filled with glial fibrils.[1]
In autopsy cases, it has been shown that white matter abnormalities are relatively confined to the cerebrum while avoiding the cerebellum and many of the major fiber tracts of the nervous system. The exception is the corticospinal tracts(pyramidal tracts) in the brainstem and sometimes spinal cord.[2]
The brain pathology of HDLS resembles that of Nasu-Hakola disease (polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy).[7]
Diagnosis
Research as of 2012 includes investigations of microglial function. This work would further clarify whether the disease is primarily a defect in microglia function. For such a study, microglial cells from HDLS kindred can be cultured from autopsy brain and analyzed in comparison to normal microglial cells on the basis of differences in mutation occurrences and growth factor expression.[5]
Differential diagnosis
Related disorders in the same disease spectrum as HDLS include Nasu-Hakola disease (polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy), and a type of leukodystrophy with pigment-filled macrophages called pigmentary orthochromatic leukodystrophy (POLD).[3] In addition to white matter disease, Nasu-Hakola causes bone cysts. It is caused by mutations in the genes involved in the same colony stimulating factor (CSF) signaling pathway cascade as identified in HDLS.[8]
Nasu-Hakola disease appears to be caused by mutations in the TYRO protein tyrosine kinase-binding protein (TYROBP - also known as DAP12) or the triggering receptor expressed on myeloid cells 2 (TREM2) protein. While different gene mutations occur within the pathway for Nasu-Hakola and HDLS, both are characterized by white matter degeneration with axonal spheroids. Current researchers in the field believe that more in depth analysis and comparison of the two genetic abnormalities in these disorders could lead to a better understanding of the disease mechanisms in these rare disorders. POLD exhibits noninflammatory demyelination of axons with initial symptoms of euphoria, apathy, headache, and executive dysfunction. While HDLS is autosomal dominant, some families with POLD have features that suggest autosomal recessive inheritance.[9] Nevertheless, POLD has recently been shown to have the same genetic basis as HDLS.
Clinical and genealogic studies
To gain a better understanding of the disease, researchers have retrospectively reviewed medical records of probands and others who were assessed through clinical examinations or questionnaires. Blood samples are collected from the families of the probands for genetic testing. These family members are assessed using their standard medical history, on their progression of Parkinson's like symptoms (Unified Parkinson's Disease Rating Scale), and on their progression of cognitive impairment such as dementia (Folstein Test).[2]
Neuroimaging
Standard MRI scans have been performed on 1.5 Tesla scanners with 5 mm thickness and 5 mm spacing to screen for white matter lesions in identified families. If signal intensities of the MRI scans are higher in white matter regions than in grey matter regions, the patient is considered to be at risk for HDLS, although a number of other disorders can also produce white matter changes and the findings are not diagnostic without genetic testing or pathologic confirmation.[2]
Pathology
Tissue sections from brain biopsies or autopsy brains are commonly embedded in paraffin from which sections are cut an mounted on glass slides for histologic studies. Special stains for myelin and axonal pathology show the abnormal changes that are characteristic of HDLS are identified in white matter of the neocortex, basal ganglia, thalamus, midbrain, pons and spinal cord.[2][10] In addition to routine histologic methods (H&E staining), samples are evaluated with immunohistochemistry for ubiquitin, amyloid precursor protein, and neurofilament to characterize axonal changes and myelin basic protein for myelin pathology. Immunohistochemical stains for microglia (CD68 or HLA-DR) and astrocytes (GFAP) are also helpful techniques to characterize white matter pathology.[6] With a similar pathology to POLD, HDLS is commonly grouped as adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP) so as to give these individually under-recognized conditions heightened attention.[3]
Classification
HDLS falls under the category of brain white matter diseases called leukoencephalopathies that are characterized by some degree of white matter dysfunction. HDLS has white matter lesions with abnormalities in myelin sheath around axons, where the causative influences are being continually explored based upon recent genetic findings. Studies by Sundal and colleagues from Sweden showed that a risk allele in Caucasians may be causative because cases identified have thus far been among large Caucasian families.[2]
Management
Epidemiology
An average clinical profile from published studies shows that the median onset age for HDLS patients is 44.3 years with a mean disease duration of 5.8 years and mean age of death at 53.2 years.[2][11] As of 2012, there have been around 15 cases identified with at least 11 sporadic cases of HDLS.[2][11] HDLS cases have been located in Germany, Norway, Sweden, and the United States, showing an international distribution focusing between Northern Europe and the United States.[2]
Through the study of numerous kindred, it was found that the disease did not occur among just males or females, but rather was evenly distributed indicative of an autosomal rather than a sex-linked genetic disorder. It was also observed that the HDLS cases did not skip generations as it would occur with a recessive inheritance, and as such has been labeled autosomal dominant.[2]
History
This disease was first described in 1984 by Axelsson et al. in a large Swedish pedigree.[12] It is a disorder better known to neuropathologists than clinicians. A neuropathologist with an interest in HDLS, Dr. Dennis W. Dickson, has identified a number of cases from neuropathology study of brains submitted for investigation of familial adult-onset dementia and movement disorders in New York and later in Florida. Recognition of the importance of this disorder as a cause of adult onset dementia and movement disorders was further heightened in 1997 at the Mayo Clinic when Dr. Zbigniew K. Wszolek identified a family with HDLS that was initially thought to be due to another disease process (FTDP-17), but only an autopsy of one and then other family members revealed it to be HDLS. Wszolek established an international consortium in 2005 to identify other families and to collect DNA or brain samples from family members for neuropathologic confirmation and genetic research at the Mayo Clinic in Florida.[2]
References
- Lin, W. L., Wszolek, Z. K., & Dickson, D. W. (2010). Hereditary diffuse leukoencephalopathy with spheroids: ultrastructural and immunoelectron microscopic studies. Int J Clin Exp Pathol, 3(7), 665-674.
- Sundal, C., Lash, J., Aasly, J., Oygarden, S., Roeber, S., Kretzschman, H., . . . Wszolek, Z. K. (2012). Hereditary diffuse leukoencephalopathy with axonal spheroids (HDLS): a misdiagnosed disease entity. J Neurol Sci, 314(1-2), 130-137. doi:10.1016/j.jns.2011.10.006
- Wider, C., Van Gerpen, J. A., DeArmond, S., Shuster, E. A., Dickson, D. W., & Wszolek, Z. K. (2009). Leukoencephalopathy with spheroids (HDLS) and pigmentary leukodystrophy (POLD): a single entity? Neurology, 72(22), 1953–1959. doi:10.1212/WNL.0b013e3181a826c0
- Rademakers, R., Baker, M., Nicholson, A., Rutherford, N., Finch, N., Soto-Ortolaza, A., . . . Wszolek, Z. (2012). Mutations in the colony stimulating factor 1 receptor (CSF1R) cause hereditary diffuse leukoencephalopathy with spheroids. Movement Disorders, 27, S399-S400.
- Kinoshita, M., Yoshida, K., Oyanagi, K., Hashimoto, T., & Ikeda, S. (2012). Hereditary diffuse leukoencephalopathy with axonal spheroids caused by R782H mutation in CSF1R: Case report. Journal of the Neurological Sciences, 318(1-2), 115-118. doi:10.1016/j.jns.2012.03.012
- Baba, Y., Ghetti, B., Baker, M. C., Uitti, R. J., Hutton, M. L., Yamaguchi, K., . . . Wszolek, Z. K. (2006). Hereditary diffuse leukoencephalopathy with spheroids: clinical, pathologic and genetic studies of a new kindred. Acta Neuropathol, 111(4), 300-311. doi:10.1007/s00401-006-0046-z
- Hancock, N., Poon, M., Taylor, B., & McLean, C. (2003). Hereditary diffuse leucoencephalopathy with spheroids. J Neurol Neurosurg Psychiatry, 74(9), 1345–1347.
- Paloneva, J., Mandelin, J., Kiialainen, A., Böhling, T., Prudlo, J., Hakola, P., . . . Peltonen, L. (2003). DAP12/TREM2 deficiency results in impaired osteoclast differentiation and osteoporotic features. The Journal of experimental medicine, 198(4), 669-675.
- Knaap, Marjo S., & Valk, Jaap. (2005). Pigmentary Orthochromatic Leukodystrophy Magnetic Resonance of Myelination and Myelin Disorders (pp. 557-558): Springer Berlin Heidelberg.
- Van Gerpen, J. A., Wider, C., Broderick, D. F., Dickson, D. W., Brown, L. A., & Wszolek, Z. K. (2008). Insights into the dynamics of hereditary diffuse leukoencephalopathy with axonal spheroids. Neurology, 71(12), 925-929. doi:10.1212/01.wnl.0000325916.30701.21
- Sundal, C., Van Gerpen, J. A., Nicholson, A. M., Wider, C., Shuster, E. A., Aasly, J., . . . Wszolek, Z. K. (2012). MRI characteristics and scoring in HDLS due to CSF1R gene mutations. Neurology, 79(6), 566-574. doi:10.1212/WNL.0b013e318263575a
- Axelsson, R., Roytta, M., Sourander, P., Akesson, H. O., & Andersen, O. (1984). Hereditary diffuse leucoencephalopathy with spheroids. Acta Psychiatr Scand Suppl, 314, 1-65.