Metachromatic leukodystrophy
Metachromatic leukodystrophy (MLD) is a lysosomal storage disease which is commonly listed in the family of leukodystrophies as well as among the sphingolipidoses as it affects the metabolism of sphingolipids. Leukodystrophies affect the growth and/or development of myelin, the fatty covering which acts as an insulator around nerve fibers throughout the central and peripheral nervous systems. MLD involves cerebroside sulfate accumulation.[1][2] Metachromatic leukodystrophy, like most enzyme deficiencies, has an autosomal recessive inheritance pattern.[2]
| Metachromatic leukodystrophy | |
|---|---|
| Other names | MLD, Arylsulfatase A deficiency, ARSA deficiency |
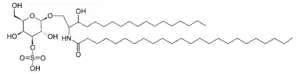 | |
| Sulfatide | |
| Specialty | Endocrinology, neurology |
| Symptoms | Progressive neurologic decline |
| Complications | Dementia, seizures, loss of motor skills |
| Usual onset | Late infantile (1-2 years), juvenile (3-20 years) or adulthood (around 40s) |
| Duration | Late infantile (3-10 years), juvenile and adult (varies) |
| Types | Late infantile, juvenile, or adult |
| Causes | Lysosomal storage disease |
| Diagnostic method | Enzyme based and genetics |
| Treatment | HSCT (pre-symptomatic), Gene therapy (late infantile), Palliative |
| Prognosis | fatal |
| Frequency | 1 in 40,000 births |
Signs and symptoms
Like many other genetic disorders that affect lipid metabolism, there are several forms of MLD, which are late infantile, juvenile, and adult.
- In the late infantile form, which is the most common form of MLD (50–60%), affected children begin having difficulty walking after the first year of life, usually at 15–24 months. Symptoms include muscle wasting and weakness, muscle rigidity, developmental delays, progressive loss of vision leading to blindness, convulsions, impaired swallowing, paralysis, and dementia. Children may become comatose. Untreated, most children with this form of MLD die by age 5, often much sooner.
- Children with the juvenile form of MLD (onset between 3 and 10 years of age) usually begin with impaired school performance, mental deterioration, and dementia, then develop symptoms similar to the late infantile form but with slower progression. Age of death is variable, but normally within 10 to 15 years of symptom onset. Some patients can live for several decades after onset. A recent trend is to try to distinguish early-juvenile (ages 3–7) and late-juvenile forms of the disease. Generally early-juveniles have motor skill declines as their first symptoms while late-juveniles show cognitive declines first.
- The adult form commonly begins after age 16 often with an onset in the 4th or 5th decade of life and presents as a psychiatric disorder or progressive dementia. Adult-onset MLD usually progresses more slowly than the late infantile and juvenile forms, with a protracted course of a decade or more.
Palliative care can help with many of the symptoms and usually improves quality of life and longevity.
Carriers have low enzyme levels compared to their family population ("normal" levels vary from family to family) but even low enzyme levels are adequate to process the body's sulfatide.
Causes
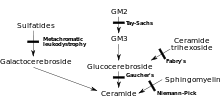
MLD is directly caused by a deficiency of the enzyme arylsulfatase A[3] (ARSA) and is characterized by enzyme activity in leukocytes that is less than 10% of normal controls.[4] However, assay of the ARSA enzyme activity alone is not sufficient for diagnosis; ARSA pseudodeficiency, which is characterized by enzyme activity that is 5~20% of normal controls does not cause MLD.[4] Without this enzyme, sulfatides build up in many tissues of the body, eventually destroying the myelin sheath of the nervous system. The myelin sheath is a fatty covering that protects nerve fibers. Without it, the nerves in the brain (central nervous system – CNS) and the peripheral nerves (peripheral nervous system – PNS) which control, among other things the muscles related to mobility, cease to function properly.
Arylsulfatase A is activated by saposin B (Sap B), a non-enzymatic proteinaceous cofactor.[5] When the arylsulfatase A enzyme level is normal but the sulfatides are still high – meaning that they are not being broken down because the enzyme is not activated – the resulting disease is saposin B deficiency, which presents similar to MLD.[4] Saposin B Deficiency is very rare, much more rare than traditional MLD.[4] The enzyme that is present is not "enabled" to a normal level of efficiency and can't break down the sulfatides which results in all of the same MLD symptoms and progression.[6]
A 2011 study contended sulfatide is not completely responsible for MLD because it is nontoxic. It has been suggested lysosulfatide, sulfatide which has had its acyl group removed, plays a role because of its cytotoxic properties in vitro.[7]
Genetics
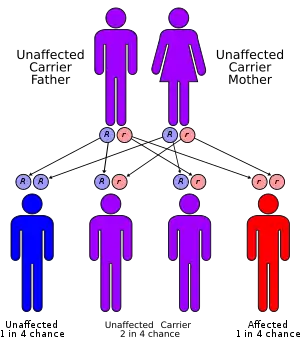
MLD has an autosomal recessive inheritance pattern. The inheritance probabilities per birth are as follows:[8]
- If both parents are carriers:
- 25% (1 in 4) children will have the disease
- 50% (2 in 4) children will be carriers, but unaffected
- 25% (1 in 4) children will be free of MLD – unaffected child that is not a carrier
- If one parent is affected and one is free of MLD:
- 0% (0) children will have the disorder – only one parent is affected, other parent always gives normal gene
- 100% (4 in 4) children will be carriers (but unaffected)
- If one parent is a carrier and the other is free of MLD:
- 50% (2 in 4) children will be carriers (but unaffected)
- 50% (2 in 4) children will be free of MLD – unaffected child that is not a carrier
In addition to these frequencies there is a 'pseudo'-deficiency that affects 7–15% of the population.[9][10] People with the pseudo deficiency do not have any MLD problems unless they also have affected status. With the current diagnostic tests, Pseudo-deficiency reports as low enzyme levels but sulfatide is processed normally so MLD symptoms do not exist. This phenomenon wreaks havoc with traditional approaches to Newborn Screening so new screening methods are being developed.
Diagnosis
Clinical examination and MRI are often the first steps in an MLD diagnosis. MRI can be indicative of MLD but is not adequate as a confirming test. An ARSA-A enzyme level blood test with a confirming urinary sulfatide test is the best biochemical test for MLD. The confirming urinary sulfatide is important to distinguish between MLD and pseudo-MLD blood results. Genomic sequencing may also confirm MLD, however, there are likely more mutations than the over 200 already known to cause MLD that are not yet ascribed to MLD that cause MLD so in those cases a biochemical test is still warranted.
Newborn screening
MLD Foundation formally launched a newborn screening initiative in late 2017. The screen development started in the early 2010s at the University of Washington Gelb Biochemistry lab. A deidentified pilot study launched in April 2016 in Washington state. Positive results led to MLD being included in the ScreenPlus identified baby research project in New York state, which is currently scheduled to launch in Q1'2021.
Treatment
There is currently no approved treatment for MLD in symptomatic late infantile patients or for juvenile and adult-onset with advanced symptoms. These patients typically receive clinical treatment focused on pain and symptom management.Pre-symptomatic late infantile MLD patients, as well as those with juvenile or adult MLD that are either presymptomatic or displaying mild symptoms, can consider bone marrow transplantation (including stem cell transplantation), which may slow down the progression of the disease in the central nervous system. However, results in the peripheral nervous system have been less dramatic, and the long-term results of these therapies have been mixed.
In 2020 the European Medical Agency, approved the Cell Therapy Drug Libmeldy for treatment of infantile and juvenile forms of metachromatic leukodystrophy in Europe. Libmeldy is a type of advanced therapy medicine called a ‘gene therapy’. This type of medicine works by delivering genes into the body. The active substance in Libmeldy is stem cells, (CD34+ cells), derived from the patient’s own bone marrow or blood, that have been modified to contain a copy of the gene to make ARSA and can divide to produce other sorts of blood cells.[11]
Atidarsagene autotemcel
A gene therapy called atidarsagene autotemcel was approved for medical use in the European Union in December 2020 and is sold under the trade name Libmeldy.[12] The indication is for use in children with the late infantile or early juvenile forms of MLD who have been identified as carriers of the defective gene but have not yet developed symptoms.[13] It is also indicated in children who have been diagnosed with the early juvenile form who have started developing symptoms but still have the ability to walk independently and before the onset of cognitive decline.[13]
In the United States, an Investigational New Drug application for atidarsagene autotemcel was accepted by the Food and Drug Administration in late 2020.[14]
Research directions
Several therapy options are currently being investigated using clinical trials primarily in late infantile patients. These therapies include gene therapy, enzyme replacement therapy (ERT), substrate reduction therapy (SRT), and potentially enzyme enhancement therapy (EET). In addition to the clinical trials, there are several other pre-clinical gene therapy research projects underway.
Epidemiology
The incidence of metachromatic leukodystrophy is estimated to occur in 1 in 40,000 to 1 in 160,000 individuals worldwide.[15] There is a much higher incidence in certain genetically isolated populations, such as 1 in 75 in Habbanites (a small group of Jews who immigrated to Israel from southern Arabia), 1 in 2,500 in the western portion of the Navajo Nation, and 1 in 8,000 among Arab groups in Israel.[15]
As an autosomal recessive disease, 1 in 40,000 equates to a 1 in 100 carrier frequency in the general population.[16]
There are an estimated 3,600 MLD births per year, with 1,900 alive in the US, 3,100 in Europe, and 49,000 alive worldwide with MLD.[16]
MLD is considered a rare disease in the US and other countries.
Research
Bone marrow and stem cell transplant therapies
- Several trials are underway to continue to improve the effectiveness and reduce the risks of bone marrow and stem cell transplants.
Gene therapy
(current as of April 2021)
Two different approaches to gene therapy are currently being researched for MLD.
- Gene therapy with an autologous stem cell transplant – Italian researchers at the San Raffaele Telethon Institute tested a novel approach combining gene therapy with an autologous stem cell transplant.[17]
- Gene therapy for late infantile and early juvenile patients was approved by the European Commission[18] in December 2020 after receiving a favorable European Medicines Agency CHMP review in October 2020.[19][20] The product is being marketed in the EU as Libmeldy. Libmeldy is a gene therapy medicinal product, for which CD34+ haematopoietic stem and progenitor cells are collected either from the patient's own bone marrow or mobilised peripheral blood.[13] These cells are transduced ex vivo using a lentiviral vector encoding the human arylsulfatase A gene to insert a functional gene to produce the ARSA enzyme.[13] When the modified cells are transplanted back into the patient as a one-time infusion, the cells have been shown to produce the missing ARSA enzyme.[13] The children by the age of five were all in good condition and going to kindergarten when normally by this age, children with the disease can not even speak.[21] Additional information can be found on the MLD Foundation's Gene Therapy page and at the Clinical Trials.gov site.
- In November, 2020, Orchard Therapeutics acknowledged IND discussions with the FDA as the part of their effort to seek FDA approval in the USA.[22]
- A trial for late juveniles was launched in February 2020.[23]
- Orchard Therapeutics acquired the gene therapy IP from GSK in April 2018.[24]
- Recruiting for the Phase I/II Clinical Trial formally started on March 24, 2010, after approval from the Italian Authorities. Recruiting the initial cohort of 8 patients was completed in mid-March 2013. The trial was to test the efficacy and safety of autologous (using the patient's own cells) hematopoietic stem cell transplantation (HSCT) after genetic modification to deliver a super-therapeutic (over-expressing) ARSA enzyme to the nervous system by the route of the blood cells. Using the patient's own stem cells with genetic correction should reduce or eliminate the complications of graft vs. host disease and provide a long-term solution to proper ARSA expression in MLD patients. Bench and animal tests showed positive results. The researchers published 2-year outcomes for the first three patients in July 2013. Results were described as promising.[21]
- The Phase I/II clinical trial is complete. No additional patients are being recruited while the data is analyzed and work progresses to improve the manufacturability and repeatability of the technology while expansion to other geographies to increase access is being considered.
- Recruiting was completed for the 20 patient cohort in April 2015, which includes an expansion in December 2014 to add 6 additional patients.
- Inclusion criteria are pre-symptomatic late infantiles and both pre- and early-symptomatic juveniles. See details on inclusion criteria and the trial protocol here.[25]
- The trial was at a single center at the San Raffaele Institute in Milan, Italy. All costs were to be paid by the researchers. This was a 3-year study. In March 2013, the last of the 8 primary trial patients started therapy. The trial had several compassionate access patients and ultimately was expanded to 20 patients
- In late 2013 GSK exercised its option for the San Rafaelle gene therapy technology and is working with the Milan Investigators to prepare for the next phase of study.[26]
- Intracerebral Gene therapy – A Phase I/II Clinical Trial started recruiting in Paris in late March, 2013 for an Intracerebral Gene Therapy clinical trial where special "vectors" carrying genetically modified material are directly injected into a dozen sites in the brain. The hope is that the corrected cells and the enzyme they produce will then diffuse into surrounding areas of the brain. Extensive work in the lab and some encouraging ALD studies provided the basis for this trial. This trial was subsequently terminated before completion.
Enzyme replacement therapy (ERT)
(current as of February 2021)
- Takeda[27] acquired the MLD ERT from Shire in early 2018[28] and continues to develop and studying their intrathecal SHP 611 (formerly HGT-1110) ERT [Enzyme Replacement Therapy].
- Clinical Trial
- A third global trial studying the late infantile form of MLD for 42 patients aged 6 – 72 months launched in April 2019 and was fully recruited in January 2021.[29] This is the first time ERT study sites are open in the US.
- Clinical trial information & inclusion criteria can be found on the MLD Foundation's ERT page and at the Clinical Trials.gov site.
Substrate reduction therapy
- Biomarin South (formerly Zacharon before being acquired by Biomarin in January 2013[30]) from San Diego had initiated a drug discovery program for MLD. This program is based on using assays which measure sulfatide accumulation in cultured fibroblasts as a means to discover and develop small molecule drugs for MLD. (This approach differs from other approaches which have measured enzyme activity to discover effective drugs.) As of July 2011, Zacharon has begun adapting the assays it developed for other lysosomal storage diseases so that they can be employed to discover and develop drugs for MLD. (current March 2013)
- The Cooper Health System (New Jersey) sponsored a clinical trial underway to determine the safety and efficacy of a Vitamin K antagonist (Warfarin) in treating Metachromatic Leukodystrophy (MLD) in 2009. No results are known to have been published.[31] (current March 2013)
Natural history studies
- A natural history study (NHS) launched in Washington, DC in January 2014 to study 30 patients with additional study centers opened in the US, Europe, South America, Southeast Asia, and South America. Due to challenges in recruiting this study has been cancelled.
- A natural history study has been underway in Pittsburgh, PA since November 2012.[32]
See also
References
- "metachromatic leukodystrophy" at Dorland's Medical Dictionary
- Le, Tao; Bhushan, Vikas; Hofmann, Jeffrey (2012). First Aid for the USMLE Step 1. McGraw-Hill. p. 117. ISBN 9780071776363.
- Poeppel P, Habetha M, Marcão A, Büssow H, Berna L, Gieselmann V (March 2005). "Missense mutations as a cause of metachromatic leukodystrophy, Degradation of arylsulfatase A in the endoplasmic reticulum". FEBS J. 272 (5): 1179–88. doi:10.1111/j.1742-4658.2005.04553.x. PMID 15720392. S2CID 9371615.
- Fluharty, Arvan. "Arylsulfatase A Deficiency: Metachromatic Leukodystrophy, ARSA Deficiency". GeneReviews, 2006
- Kishimoto Y, Hiraiwa M, O'Brien JS (Sep 1992). "Saposins: structure, function, distribution, and molecular genetics". J. Lipid Res. 33 (9): 1255–67. doi:10.1016/S0022-2275(20)40540-1. PMID 1402395.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - "Genetics". MLD Foundation. Archived from the original on 2014-12-22. Retrieved 2017-05-28.
- Blomqvist, M.; Gieselmann, V.; Månsson, J. E. (2011). "Accumulation of lysosulfatide in the brain of arylsulfatase A-deficient mice". Lipids in Health and Disease. 10 (1): 28. doi:10.1186/1476-511X-10-28. PMC 3041674. PMID 21299873.
- "Autosomal recessive: MedlinePlus Medical Encyclopedia". medlineplus.gov. Retrieved 2021-08-18.
- Hohenschutz, C; Eich P; Friedl W; Waheed A; Conzelmann E; Propping P. (April 1989). "Pseudodeficiency of arylsulfatase A". Human Genetics. 82 (1): 45–8. doi:10.1007/bf00288270. PMID 2565866. S2CID 32274162.
- Herz, Barbara; Bach, G. (1984). "Arylsulfatase A in pseudodeficiency". Human Genetics. 66 (2–3): 147–150. doi:10.1007/BF00286589. PMID 6143719. S2CID 2349721.
- "Libmeldy", ema.europa.eu
- "Libmeldy EPAR". European Medicines Agency (EMA). Retrieved 22 December 2020.
- "New gene therapy to treat rare genetic disorder metachromatic leukodystrophy". European Medicines Agency (EMA) (Press release). 16 October 2020. Retrieved 16 October 2020. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- "Orchard Therapeutics Announces FDA Clearance of IND Application for OTL-200 for Metachromatic Leukodystrophy (MLD) | Orchard Therapeutics". ir.orchard-tx.com. Retrieved 2021-04-29.
- Metachromatic leukodystrophy at Genetics Home Reference. Reviewed September 2007
- "MLD 101: Genetics". www.mldfoundation.org. January 6, 2017. Archived from the original on December 30, 2013. Retrieved January 6, 2017.
- Biffi A, Lucchini G, Rovelli A, Sessa M (October 2008). "Metachromatic leukodystrophy: an overview of current and prospective treatments". Bone Marrow Transplant. 42 Suppl 2: S2–6. doi:10.1038/bmt.2008.275. PMID 18978739.
- Orchard, Therapeutics (21 December 2020). "Orchard Therapeutics Receives EC Approval for Libmeldy for the Treatment of Early-Onset Metachromatic Leukodystrophy (MLD)". Intrado. GlobalNewsWire. Retrieved 12 January 2021.
- Orchard, Therapeutics. "Orchard Therapeutics Receives Positive CHMP Opinion for Libmeldy for the Treatment of Early-Onset Metachromatic Leukodystrophy (MLD)". No. 16 October 2020. Retrieved 12 January 2021.
- American, Pharmaceutical Review. "Orchard Therapeutics Announces MAA Filing of Metachromatic Leukodystrophy Treatment". American Pharmaceutical Review. CompareNetworks. Retrieved 3 December 2019.
- Biffi A, Montini E, Lorioli L, et al. (2013). "Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy". Science. 341 (6148): 1233158. doi:10.1126/science.1233158. PMID 23845948. S2CID 206546808.
- Orchard, Therapeutics (19 November 2020). "Orchard Therapeutics Announces FDA Clearance of IND Application for OTL-200 for Metachromatic Leukodystrophy (MLD)". Intrado. Global NewWire. Retrieved 12 January 2021.
- "OTL-200 in Patients With Late Juvenile Metachromatic Leukodystrophy (MLD)". ClinicalTrials.Gov. Retrieved 25 February 2020.
- Orchard, Therapeutics (12 April 2018). "GSK signs strategic agreement to transfer rare disease gene therapy portfolio to Orchard Therapeutics". Retrieved 12 January 2021.
- "MLD gene therapy - San Raffaele - MLD Foundation". MLD Foundation.
- "GSK Product Pipeline". GSK. March 2014. Retrieved 29 June 2014.
- "Takeda Pipeline)". Takeda Pipeline. Retrieved 12 September 2020.
- "Takeda Completes Acquisition of Shire, Becoming a Global, Values-based, R&D-Driven Biopharmaceutical Leader". Takeda.com. Retrieved 7 January 2018.
- "A Study of Intrathecal SHP611 in Participants With Late Infantile Metachromatic Leukodystrophy (Embolden)". ClinicalTrails.gov. Retrieved 30 April 2019.
- "BioMarin Acquires Zacharon Pharmaceuticals (NASDAQ:BMRN)". Archived from the original on 2013-01-29. Retrieved 2013-03-16.
- "Effect of Warfarin in the Treatment of Metachromatic Leukodystrophy - Full Text View - ClinicalTrials.gov". clinicaltrials.gov. 18 March 2011.
- "NDRD: Program for the Study of Neurodevelopment in Rare Disorders". NDRD: Program for the Study of Neurodevelopment in Rare Disorders. Retrieved 12 September 2020.
- Some portions of this article are courtesy of the public domain text available at the National Institute of Neurological Disorders and Stroke:
- "NINDS Metachromatic Leukodystrophy Information Page". Retrieved 2009-06-07.