Spinal muscular atrophy
Spinal muscular atrophy (SMA) is a rare neuromuscular disorder that results in the loss of motor neurons and progressive muscle wasting.[3][4][5] It is usually diagnosed in infancy or early childhood and if left untreated it is the most common genetic cause of infant death.[6] It may also appear later in life and then have a milder course of the disease. The common feature is progressive weakness of voluntary muscles, with arm, leg and respiratory muscles being affected first.[7][8] Associated problems may include poor head control, difficulties swallowing, scoliosis, and joint contractures.[2][8]
| Spinal muscular atrophy | |
|---|---|
| Other names | Autosomal recessive proximal spinal muscular atrophy, 5q spinal muscular atrophy |
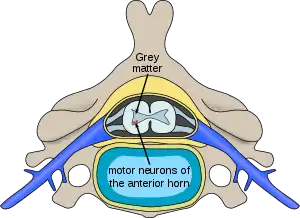 | |
| Location of neurons affected by spinal muscular atrophy in the spinal cord | |
| Specialty | Neurology |
| Symptoms | Progressive muscle weakness[1] |
| Complications | Scoliosis, joint contractures, pneumonia[2] |
| Usual onset | Mutation is congenital, symptoms start varies by type |
| Duration | Lifelong |
| Types | Type 0 to type 4[2] |
| Causes | Mutation in SMN1[2] |
| Diagnostic method | Genetic testing[1] |
| Differential diagnosis | Congenital muscular dystrophy, Duchenne muscular dystrophy, Prader-Willi syndrome[2] |
| Treatment | Supportive care, medications[1] |
| Medication | Nusinersen, onasemnogene abeparvovec, Risdiplam |
| Prognosis | Varies by type[2] |
| Frequency | 1 in 10,000 people[2] |
The age of onset and the severity of symptoms form the basis of the traditional classification of spinal muscular atrophy into a number of types.[4]
Spinal muscular atrophy is due to an abnormality (mutation) in the SMN1 gene[1][2] which encodes SMN, a protein necessary for survival of motor neurons.[8] Loss of these neurons in the spinal cord prevents signalling between the brain and skeletal muscles.[8] Another gene, SMN2, is considered a disease modifying gene, since usually the more the SMN2 copies, the milder is the disease course. The diagnosis of SMA is based on symptoms and confirmed by genetic testing.[9][1]
Usually, the mutation in the SMN1 gene is inherited from both parents in an autosomal recessive manner, although in around 2% of cases it occurs during early development (de novo).[1][10] The incidence of spinal muscular atrophy worldwide varies from about 1 in 4,000 births to around 1 in 16,000 births,[11] with 1 in 7,000 and 1 in 10,000 commonly quoted for Europe and the US respectively.[2]
Outcomes in the natural course of the disease vary from death within a few weeks after birth in the most acute cases to normal life expectancy in the protracted SMA forms.[8] The introduction of causative treatments in 2016 has significantly improved the outcomes. Medications that target the genetic cause of the disease include nusinersen, risdiplam, and the gene therapy medication onasemnogene abeparvovec. Supportive care includes physical therapy, occupational therapy, respiratory support, nutritional support, orthopaedic interventions, and mobility support.[1]
Classification
5q SMA is a single disease that manifests over a wide range of severity, affecting infants through adults. Before its genetics was understood, its varying manifestations were thought to be different diseases – Werdnig–Hoffmann disease when young children were affected and Kugelberg–Welander disease for late-onset cases.[12]
In 1990, it was realised that these separate diseases formed a spectrum of the same disorder. Spinal muscular atrophy was then classified into 3–5 clinical types based either on the age of symptom onset or on the maximum motor function achieved.[10][12] Currently, the consensus is that the phenotype of spinal muscular atrophy spans a continuum of symptoms without clear delineation of subtypes.[10] However, the traditional classification, outlined in the table below, is still used today both in clinical research and sometimes, controversially, as a criterion of access to therapies.
| Type | Eponym | Usual age of onset | Natural history (without pharmacological treatment) | OMIM |
|---|---|---|---|---|
| SMA 0 | Prenatal | Symptoms are observed at birth and often become apparent in the prenatal period as reduced foetal movement. Affected children typically have only a single copy of the SMN2 gene and usually survive only a few weeks even with 24/7 respiratory support. This form is very rare – accounts for approx. 2% of cases. | ||
| SMA 1 (Infantile) |
Werdnig–Hoffmann disease | 0–6 months | This form is diagnosed in around 50% of patients, in whom the disease manifests in the first few weeks or months of life. SMA then has a quick and unexpected onset, with various muscle groups failing progressively. Infants never learn to sit unsupported and most gradually lose most of their muscle function. Death is usually caused by the failure of the respiratory muscles induced by pneumonia (frequently, aspiration pneumonia). Unless offered respiratory support and/or pharmacological treatment early, babies diagnosed with SMA type 1 do not generally survive past two years of age. With proper respiratory support, those with milder SMA type 1 phenotypes, which account for around 10% of SMA 1 cases, are known to survive into adolescence and adulthood even without pharmacological treatment, although they always require round-the-clock care. | 253300 |
| SMA 2 (Intermediate) |
Dubowitz disease | 6–18 months | The intermediate form, diagnosed in around 20% of patients, denotes people who were able to maintain a sitting position at least some time in their life but never learned to walk unsupported. The onset of weakness is usually noticed some time between 6 and 18 months of life. The progress is known to vary greatly, some people gradually grow weaker over time while others through careful maintenance remain relatively stable. Body muscles are weakened, and the respiratory system is a major concern as are muscle contractures and spinal curvature. Life expectancy is reduced, even as most people with SMA 2 live well into adulthood even without treatment. | 253550 |
| SMA 3 (Juvenile) |
Kugelberg–Welander disease | >12 months | The juvenile form, diagnosed in around 30% of patients, manifests after 12 months of age, or after the children have already learned to make at least a few independent steps. The disease progresses slowly, and most people with SMA 3 lose walking ability sometime in their lives, requiring mobility support. Respiratory involvement is rare and life expectancy is normal or near-normal. | 253400 |
| SMA 4 (Adult onset) |
Adulthood | This denotes the adult-onset form, sometimes also classified as a late-onset SMA type 3. It occurs in approx. 5% of patients and usually manifests in the third or fourth decade of life. The symptoms consist of gradual weakening of leg muscles, which frequently makes it necessary for the patient to use walking aids. Other complications are rare and life expectancy is unaffected. | 271150 |
For convenience, care-focused publications classify patients into "non-sitters", "sitters" and "walkers" based on their actual functional status.
Motor development and disease progression in people with SMA is usually assessed using validated functional scales – CHOP-INTEND (The Children's Hospital of Philadelphia Infant Test of Neuromuscular Disorders) or HINE (Hammersmith Infant Neurological Examination) in infants; and either the MFM (Motor Function Measure) or one of several variants of the HFMS (Hammersmith Functional Motor Scale)[13][14][15][16] in older patients.
The eponymous label Werdnig–Hoffmann disease (sometimes misspelled with a single n) refers to the earliest clinical descriptions of childhood SMA by Johann Hoffmann and Guido Werdnig.[12] The eponymous term Kugelberg–Welander disease named after Erik Klas Hendrik Kugelberg (1913–1983) and Lisa Welander (1909–2001), who first documented the late-onset form and distinguished it from muscular dystrophy.[12] Very rarely used Dubowitz disease (not to be confused with Dubowitz syndrome) is named after Victor Dubowitz, an English neurologist who authored several studies on the intermediate SMA phenotype.
Signs and symptoms
The symptoms vary depending on the SMA type, the stage of the disease as well as individual factors. Signs and symptoms below are most common in the severe SMA type 0/I:[17]
- Areflexia, particularly in extremities
- Overall muscle weakness, poor muscle tone, limpness or a tendency to flop
- Difficulty achieving developmental milestones, difficulty sitting/standing/walking
- In small children: adopting of a frog-leg position when sitting (hips abducted and knees flexed)
- Loss of strength of the respiratory muscles: weak cough, weak cry (infants), accumulation of secretions in the lungs or throat, respiratory distress
- Bell-shaped torso (caused by using only abdominal muscles for respiration) in severe SMA type
- Fasciculations (twitching) of the tongue
- Difficulty sucking or swallowing, poor feeding
Causes

Spinal muscular atrophy is caused by a genetic mutation in the SMN1 gene.[18]
Human chromosome 5 contains two nearly identical genes at location 5q13: a telomeric copy SMN1 and a centromeric copy SMN2. In healthy individuals, the SMN1 gene codes the survival of motor neuron protein (SMN) which, as its name says, plays a crucial role in survival of motor neurons. The SMN2 gene, on the other hand – due to a variation in a single nucleotide (840.C→T) – undergoes alternative splicing at the junction of intron 6 to exon 8, with only 10–20% of SMN2 transcripts coding a fully functional survival of motor neuron protein (SMN-fl) and 80–90% of transcripts resulting in a truncated protein compound (SMNΔ7) which is rapidly degraded in the cell.[19]
In individuals affected by SMA, the SMN1 gene is mutated in such a way that it is unable to correctly code the SMN protein – due to either a deletion[20] occurring at exon 7[21] or to other point mutations (frequently resulting in the functional conversion of the SMN1 sequence into SMN2). Almost all people, however, have at least one functional copy of the SMN2 gene (with most having 2–4 of them) which still codes 10–20% of the usual level of the SMN protein, allowing some neurons to survive. In the long run, however, the reduced availability of the SMN protein results in gradual death of motor neuron cells in the anterior horn of spinal cord and the brain. Skeletal muscles, which all depend on these motor neurons for neural input, now have decreased innervation (also called denervation), and therefore have decreased input from the central nervous system (CNS). Decreased impulse transmission through the motor neurons leads to decreased contractile activity of the denervated muscle. Consequently, denervated muscles undergo progressive atrophy (waste away).
Muscles of lower extremities are usually affected first, followed by muscles of upper extremities, spine and neck and, in more severe cases, pulmonary and mastication muscles. Proximal muscles are always affected earlier and to a greater degree than distal muscles.[22]
The severity of SMA symptoms is broadly related to how well the remaining SMN2 genes can make up for the loss of function of SMN1. This partly depends on the number of copies of the SMN2 gene present on the chromosome. Whilst healthy individuals usually carry two SMN2 gene copies, people with SMA can have anything between 1 and 5 (or more) of them; the greater the number of SMN2 copies, the milder the disease severity. Thus, most SMA type I babies have one or two SMN2 copies; people with SMA II and III usually have at least three SMN2 copies; and people with SMA IV normally have at least four of them. However, the correlation between symptom severity and SMN2 copy number is not absolute and there seem to exist other factors affecting the disease phenotype.[23]
Spinal muscular atrophy is inherited in an autosomal recessive pattern, which means that the defective gene is located on an autosome. Two copies of the defective gene – one from each parent – are required to inherit the disorder: the parents may be carriers and not personally affected. SMA seems to appear de novo (i.e., without any hereditary causes) in around 2–4% of cases.
Spinal muscular atrophy affects individuals of all ethnic groups, unlike other well known autosomal recessive disorders, such as sickle cell disease and cystic fibrosis, which have significant differences in occurrence rate among ethnic groups. The overall prevalence of SMA, of all types and across all ethnic groups, is in the range of 1 per 10,000 individuals; the gene frequency is around 1:100, therefore, approximately one in 50 persons are carriers.[24][25] There are no known health consequences of being a carrier. A person may learn carrier status only if one's child is affected by SMA or by having the SMN1 gene sequenced.
Affected siblings usually have a very similar form of SMA. However, occurrences of different SMA types among siblings do exist – while rare, these cases might be due to additional de novo deletions of the SMN gene, not involving the NAIP gene, or the differences in SMN2 copy numbers.
Diagnosis
SMA is diagnosed using genetic testing that detects homozygous deletion of the SMN1 gene in over 95% of cases,[17] and a compound SMN1 mutation in the remaining patients. Genetic testing is usually carried out using a blood sample, and MLPA is one of more frequently used genetic testing techniques, as it also allows establishing the number of SMN2 gene copies, which has clinical importance.[17]
Symptomatically, SMA can be diagnosed with a degree of certainty only in children with the acute form who manifest a progressive illness with paradoxical breathing, bilateral low muscle tone and absent tendon reflexes.
Early diagnosis
Early diagnosis of SMA, at the asymptomatic stage of the disease, allows for
Preimplantation testing
Preimplantation genetic diagnosis can be used to screen for SMA-affected embryos during in-vitro fertilisation.
Prenatal testing
Prenatal testing for SMA is possible through chorionic villus sampling, cell-free fetal DNA analysis and other methods.
Newborn screening
Routine newborn screening for SMA is becoming increasingly commonplace in developed countries, given the availability of causative treatments that are most effective at the asymptomatic stage of the disease.[26][27][28] In 2018, newborn screening for SMA was added to the US list of recommended newborn screening tests[29][30][31] and as of April 2020 it has been adopted in 39 US states.[32][33] As of May 2021, SMA newborn screening has been implemented in Taiwan[34] and is in the course of implementation in Australia,[35] Belgium,[36] Canada, France, Germany,[37] Netherlands,[38] Poland, Serbia and Slovenia. Additionally, pilot projects are being conducted in Australia, China,[39] Italy, and Japan.[40]
Carrier testing
Those at risk of being carriers of SMN1 deletion, and thus at risk of having offspring affected by SMA, can undergo carrier analysis using a blood or saliva sample. The American College of Obstetricians and Gynecologists recommends all people thinking of becoming pregnant be tested to see if they are a carrier.[41] The carrier frequency of SMA is comparable to other disorders like thalassemia and in a north Indian cohort has been found to be 1 in 38.[42] However, genetic testing will not be able to identify all individuals at risk since about 2% of cases are caused by de novo mutations and 5% of the normal population have two copies of SMN1 on the same chromosome, which makes it possible to be a carrier by having one chromosome with two copies and a second chromosome with zero copies. This situation will lead to a false negative result, as the carrier status will not be correctly detected by a traditional genetic test.[43][44]
Management
The management of SMA varies based upon the severity and type. In the most severe forms (types 0/1), individuals have the greatest muscle weakness requiring prompt intervention. Whereas the least severe form (type 4/adult onset), individuals may not seek the certain aspects of care until later (decades) in life. While types of SMA and individuals among each type may differ, therefore specific aspects of an individual's care can differ.
Medication
Nusinersen (marketed as Spinraza) is used to treat spinal muscular atrophy.[45] It is an antisense nucleotide that modifies the alternative splicing of the SMN2 gene.[45] It is given directly to the central nervous system using an intrathecal injection.[45][46] Nusinersen prolongs survival and improves motor function in infants with SMA.[47][48] It was approved for use in the US in 2016, and for use in the EU in 2017.[49][50][51]
Onasemnogene abeparvovec (marketed as Zolgensma) is a gene therapy treatment which uses self-complementary adeno-associated virus type 9 (scAAV-9) as a vector to deliver the SMN1 transgene.[52][53] The therapy was first approved in the US in May 2019 as an intravenous formulation for children below 24 months of age.[54][55] Approval in the European Union, Japan and other countries followed, albeit often with different approval scopes.[56][57]
Risdiplam (marketed as Evrysdi) is a medication taken by mouth in liquid form.[58][59] It is a pyridazine derivative that works by increasing the amount of functional survivor motor neuron protein produced by the SMN2 gene through modifying its splicing pattern.[60][61] Risdiplam was first approved for medical use in the United States in August 2020[58] and has since been approved in over 30 countries.
Breathing
The respiratory system is the most common system to be affected and the complications are the leading cause of death in SMA types 0/1 and 2. SMA type 3 can have similar respiratory problems, but it is more rare.[22] The complications that arise due to weakened intercostal muscles because of the lack of stimulation from the nerve. The diaphragm is less affected than the intercostal muscles.[22] Once weakened, the muscles never fully recover the same functional capacity to help in breathing and coughing as well as other functions. Therefore, breathing is more difficult and pose a risk of not getting enough oxygen/shallow breathing and insufficient clearance of airway secretions. These issues more commonly occurs while asleep, when muscles are more relaxed. Swallowing muscles in the pharynx can be affected, leading to aspiration coupled with a poor coughing mechanism increases the likelihood of infection/pneumonia.[62] Mobilizing and clearing secretions involve manual or mechanical chest physiotherapy with postural drainage, and manual or mechanical cough assistance device. To assist in breathing, Non-invasive ventilation (BiPAP) is frequently used and tracheostomy may be sometimes performed in more severe cases;[63] both methods of ventilation prolong survival to a comparable degree, although tracheostomy prevents speech development.[64]
Nutrition
The more severe the type of SMA, the more likely to have nutrition related health issues. Health issues can include difficulty in feeding, jaw opening, chewing and swallowing. Individuals with such difficulties can be at increase risk of over or undernutrition, failure to thrive and aspiration. Other nutritional issues, especially in individuals that are non-ambulatory (more severe types of SMA), include food not passing through the stomach quickly enough, gastric reflux, constipation, vomiting and bloating.[65] Therein, it could be necessary in SMA type I and people with more severe type II to have a feeding tube or gastrostomy.[65][66][67] Additionally, metabolic abnormalities resulting from SMA impair β-oxidation of fatty acids in muscles and can lead to organic acidemia and consequent muscle damage, especially when fasting.[68][69] It is suggested that people with SMA, especially those with more severe forms of the disease, reduce intake of fat and avoid prolonged fasting (i.e., eat more frequently than healthy people)[70] as well as choosing softer foods to avoid aspiration.[62] During an acute illness, especially in children, nutritional problems may first present or can exacerbate an existing problem (example: aspiration) as well as cause other health issues such as electrolyte and blood sugar disturbances.[71]
Orthopaedics
Skeletal problems associated with weak muscles in SMA include tight joints with limited range of movement, hip dislocations, spinal deformity, osteopenia, an increase risk of fractures and pain.[22] Weak muscles that normally stabilize joints such as the vertebral column lead to development of kyphosis and/or scoliosis and joint contracture.[22] Spine fusion is sometimes performed in people with SMA I/II once they reach the age of 8–10 to relieve the pressure of a deformed spine on the lungs. Furthermore, immobile individuals, posture and position on mobility devices as well as range of motion exercises, and bone strengthening can be important to prevent complications.[71] People with SMA might also benefit greatly from various forms of physiotherapy, occupational therapy and physical therapy.
Orthotic devices can be used to support the body and to aid walking. For example, orthotics such as AFOs (ankle foot orthoses) are used to stabilise the foot and to aid gait, TLSOs (thoracic lumbar sacral orthoses) are used to stabilise the torso. Assistive technologies may help in managing movement and daily activity and greatly increase the quality of life.
Other
Although the heart is not a matter of routine concern, a link between SMA and certain heart conditions has been suggested.[72][73][74][75]
Children with SMA do not differ from the general population in their behaviour; their cognitive development can be slightly faster, and certain aspects of their intelligence are above the average.[76][77][78] Despite their disability, SMA-affected people report high degree of satisfaction from life.[79]
Palliative care in SMA has been standardised in the Consensus Statement for Standard of Care in Spinal Muscular Atrophy[22] which has been recommended for standard adoption worldwide.
Prognosis
In the absence of pharmacological treatment, people with SMA tend to deteriorate over time. Recently, survival has increased in severe SMA patients with aggressive and proactive supportive respiratory and nutritional support.[80]
If left untreated, the majority of children diagnosed with SMA type 0 and 1 do not reach the age of 4, recurrent respiratory problems being the primary cause of death.[81] With proper care, milder SMA type I cases (which account for approx. 10% of all SMA1 cases) live into adulthood.[82] Long-term survival in SMA type I is not sufficiently evidenced; however, as of 2007 advances in respiratory support seem to have brought down mortality.[83]
In untreated SMA type II, the course of the disease is slower to progress and life expectancy is less than the healthy population. Death before the age of 20 is frequent, although many people with SMA live to become parents and grandparents. SMA type III has normal or near-normal life expectancy if standards of care are followed. Type IV, adult-onset SMA usually means only mobility impairment and does not affect life expectancy.
Research directions
Since the underlying genetic cause of SMA was identified in 1995,[20] several therapeutic approaches have been proposed and investigated that primarily focus on increasing the availability of SMN protein in motor neurons.[84] The main research directions have been as follows:
SMN1 gene replacement
Gene therapy in SMA aims at restoring the SMN1 gene function through inserting specially crafted nucleotide sequence (a SMN1 transgene) into the cell nucleus using a viral vector. This approach has been exploited by the first approved gene therapy for SMA, scAAV-9 based treatment onasemnogene abeparvovec.[85]
SMN2 alternative splicing modulation
This approach aims at modifying the alternative splicing of the SMN2 gene to force it to code for higher percentage of full-length SMN protein. Sometimes it is also called gene conversion, because it attempts to convert the SMN2 gene functionally into SMN1 gene. It is the therapeutic mechanism of the approved medications nusinersen and risdiplam.
Branaplam is another SMN2 splicing modulator that has reached the clinical stage of development.[86]
Historically, this research direction investigated also other molecules. RG3039, also known as Quinazoline495, was a proprietary quinazoline derivative developed by Repligen and licensed to Pfizer in March 2014 which was discontinued shortly after, having only completed phase I trials. PTK-SMA1 was a proprietary small-molecule splicing modulator of the tetracyclines group developed by Paratek Pharmaceuticals and about to enter clinical development in 2013 which however never happened due to Paratek downsizing at that time. RG7800, developed by Hoffmann-La Roche, was a molecule akin to risdiplam that has undergone phase I testing but was discontinued due to animal toxicity.[87] Early leads also included sodium orthovanadate[88] and aclarubicin.[89]
Morpholino-type antisense oligonucleotides, with the same cellular target as nusinersen, remain a subject of research in treating SMA and other single-gene diseases, including at the University College London[90] and at the University of Oxford.[91]
SMN2 gene activation
This approach aims at increasing expression (activity) of the SMN2 gene, thus increasing the amount of full-length SMN protein available.
- Oral salbutamol (albuterol), a popular asthma medicine, showed therapeutic potential in SMA both in vitro[92] and in three small-scale clinical trials involving patients with SMA types 2 and 3,[93][94][95] besides offering respiratory benefits.
A few compounds initially showed promise but failed to demonstrate efficacy in clinical trials. Butyrates (sodium butyrate and sodium phenylbutyrate) held some promise in in vitro studies[96][97][98] but a clinical trial in symptomatic people did not confirm their efficacy.[99] Another clinical trial in pre-symptomatic types 1–2 infants was completed in 2015 but no results have been published.[100]
- Valproic acid (VPA) was used in SMA on an experimental basis in the 1990s and 2000s because in vitro research suggested its moderate effectiveness.[101][102] However, it demonstrated no efficacy in achievable concentrations when subjected to a large clinical trial.[103][104][105] It has also been proposed that it may be effective in a subset of people with SMA but its action may be suppressed by fatty acid translocase in others.[106] Others argue it may actually aggravate SMA symptoms.[107] It is currently not used due to the risk of severe side effects related to long-term use. A 2019 meta-analysis suggested that VPA may offer benefits, even without improving functional score.[108]
- Hydroxycarbamide (hydroxyurea) was shown effective in mouse models[109] and subsequently commercially researched by Novo Nordisk, Denmark, but demonstrated no effect on people with SMA in subsequent clinical trials.[110]
Compounds which increased SMN2 activity in vitro but did not make it to the clinical stage include growth hormone, various histone deacetylase inhibitors,[111] benzamide M344,[112] hydroxamic acids (CBHA, SBHA, entinostat, panobinostat,[113] trichostatin A,[114][115] vorinostat[116]), prolactin[117] as well as natural polyphenol compounds like resveratrol and curcumin.[118][119] Celecoxib, a p38 pathway activator, is sometimes used off-label by people with SMA based on a single animal study[120] but such use is not backed by clinical-stage research.
SMN stabilisation
SMN stabilisation aims at stabilising the SMNΔ7 protein, the short-lived defective protein coded by the SMN2 gene, so that it is able to sustain neuronal cells.[121]
No compounds have been taken forward to the clinical stage. Aminoglycosides showed capability to increase SMN protein availability in two studies.[122][123] Indoprofen offered some promise in vitro.[124]
Neuroprotection
Neuroprotective drugs aim at enabling the survival of motor neurons even with low levels of SMN protein.
- Olesoxime was a proprietary neuroprotective compound developed by the French company Trophos, later acquired by Hoffmann-La Roche, which showed stabilising effect in a phase-II clinical trial involving people with SMA types 2 and 3. Its development was discontinued in 2018 in view of competition from nusinersen and underwhelming data from an open-label extension trial.[125]
Of clinically studied compounds which did not show efficacy, thyrotropin-releasing hormone (TRH) held some promise in an open-label uncontrolled clinical trial[126][127][128] but did not prove effective in a subsequent double-blind placebo-controlled trial.[129] Riluzole, a drug that offers limited clinical benefit in amyotrophic lateral sclerosis, was proposed to be similarly tested in SMA;[130][131] however, a 2008–2010 trial in SMA types 2 and 3[132] was stopped early due to the lack of satisfactory results.[133] Other compounds that displayed some neuroprotective effect in in vitro research but never moved on to in vivo studies include β-lactam antibiotics (e.g., ceftriaxone)[134][135] and follistatin.[136]
Muscle restoration
This approach aims to counter the effect of SMA by targeting the muscle tissue instead of neurons.
- Reldesemtiv (CK-2127107, CK-107) is a skeletal troponin activator developed by Cytokinetics in cooperation with Astellas. The drug aims at increasing muscle reactivity despite lowered neural signalling. The molecule showed some success in phase II clinical trial in adolescent and adults with SMA types 2, 3, and 4.[137]
- Apitegromab (SRK-015) is monoclonal antibody that blocks the activation of the skeletal muscle protein myostatin, thereby promoting muscle tissue growth. As of 2021, the molecule showed success as an experimental add-on treatment in paediatric and adult patients treated with nusinersen.[138]
- GYM329 (RO7204239), developed by Hoffman-La Roche, works similarly to apitegromab by blocking myostatin activation. As of 2022, it is undergoing clinical development in non-ambulant children with SMA aged 2–10, combined with risdiplam.[139]
Stem cells
Whilst stem cells never form a part of any recognised therapy for SMA, a number of private companies, usually located in countries with lax regulatory oversight, take advantage of media hype and market stem cell injections as a "cure" for a vast range of disorders, including SMA. The medical consensus is that such procedures offer no clinical benefit whilst carrying significant risk, therefore people with SMA are advised against them.[140][141] In 2013–2014, a small number of SMA1 children in Italy received court-mandated stem cell injections following the Stamina scam, but the treatment was reported having no effect[142][143]
Registries
People with SMA in the European Union can participate in clinical research by entering their details into registries managed by TREAT-NMD.[144]
See also
- Accomable
- Motor neuron disease
References
- "Spinal muscular atrophy". Genetic and Rare Diseases Information Center (GARD) – an NCATS Program. Retrieved 27 May 2019.
- "Spinal Muscular Atrophy". NORD (National Organization for Rare Disorders). Retrieved 27 May 2019.
- "Spinal muscular atrophy". nhs.uk. 23 October 2017. Retrieved 24 October 2020.
- "Spinal muscular atrophy: MedlinePlus Genetics". medlineplus.gov. Retrieved 24 October 2020.
- "Spinal Muscular Atrophy (SMA) | Boston Children's Hospital". www.childrenshospital.org. Retrieved 25 October 2020.
- "FDA approves innovative gene therapy to treat pediatric patients with spinal muscular atrophy, a rare disease and leading genetic cause of infant mortality". FDA. 24 May 2019. Retrieved 27 May 2019.
- "Spinal Muscular Atrophy Fact Sheet | National Institute of Neurological Disorders and Stroke". NINDS. Retrieved 27 May 2019.
- "Spinal muscular atrophy". Genetics Home Reference. Retrieved 27 May 2019.
- "Spinal Muscular Atrophy – Conditions | Children's National". childrensnational.org. Retrieved 25 October 2020.
- Prior, Thomas W.; Leach, Meganne E.; Finanger, Erika (1993), Adam, Margaret P.; Ardinger, Holly H.; Pagon, Roberta A.; Wallace, Stephanie E. (eds.), "Spinal Muscular Atrophy", GeneReviews®, Seattle (WA): University of Washington, Seattle, PMID 20301526, retrieved 25 October 2020
- Verhaart, Ingrid E. C.; Robertson, Agata; Leary, Rebecca; McMacken, Grace; König, Kirsten; Kirschner, Janbernd; Jones, Cynthia C.; Cook, Suzanne F.; Lochmüller, Hanns (July 2017). "A multi-source approach to determine SMA incidence and research ready population". Journal of Neurology. 264 (7): 1465–1473. doi:10.1007/s00415-017-8549-1. ISSN 0340-5354. PMC 5502065. PMID 28634652.
- Dubowitz, Victor (2009). "Ramblings in the history of spinal muscular atrophy". Neuromuscular Disorders. 19 (1): 69–73. doi:10.1016/j.nmd.2008.10.004. PMID 18951794. S2CID 37576912.
- Main M, Kairon H, Mercuri E, Muntoni F (2003). "The Hammersmith functional motor scale for children with spinal muscular atrophy: a scale to test ability and monitor progress in children with limited ambulation". European Journal of Paediatric Neurology. 7 (4): 155–9. doi:10.1016/S1090-3798(03)00060-6. PMID 12865054.
- Krosschell KJ, Maczulski JA, Crawford TO, Scott C, Swoboda KJ (July 2006). "A modified Hammersmith functional motor scale for use in multi-center research on spinal muscular atrophy". Neuromuscular Disorders. 16 (7): 417–26. doi:10.1016/j.nmd.2006.03.015. PMC 3260054. PMID 16750368.
- O'Hagen JM, Glanzman AM, McDermott MP, Ryan PA, Flickinger J, Quigley J, Riley S, Sanborn E, Irvine C, Martens WB, Annis C, Tawil R, Oskoui M, Darras BT, Finkel RS, De Vivo DC (October 2007). "An expanded version of the Hammersmith Functional Motor Scale for SMA II and III patients". Neuromuscular Disorders. 17 (9–10): 693–7. doi:10.1016/j.nmd.2007.05.009. PMID 17658255. S2CID 10365924.
- Glanzman AM, O'Hagen JM, McDermott MP, Martens WB, Flickinger J, Riley S, Quigley J, Montes J, Dunaway S, Deng L, Chung WK, Tawil R, Darras BT, De Vivo DC, Kaufmann P, Finkel RS, et al. (Pediatric Neuromuscular Clinical Research Network for Spinal Muscular Atrophy (PNCR)) (December 2011). "Validation of the Expanded Hammersmith Functional Motor Scale in spinal muscular atrophy type II and III". Journal of Child Neurology. 26 (12): 1499–507. doi:10.1177/0883073811420294. PMID 21940700. S2CID 206549483.
- Oskoui M, Darras BT, DeVivo DC (2017). "Chapter 1". In Sumner CJ, Paushkin S, Ko CP (eds.). Spinal Muscular Atrophy: Disease Mechanisms. Elsevier. ISBN 978-0-12-803685-3.
- Brzustowicz LM, Lehner T, Castilla LH, Penchaszadeh GK, Wilhelmsen KC, Daniels R, Davies KE, Leppert M, Ziter F, Wood D (April 1990). "Genetic mapping of chronic childhood-onset spinal muscular atrophy to chromosome 5q11.2–13.3". Nature. 344 (6266): 540–1. Bibcode:1990Natur.344..540B. doi:10.1038/344540a0. PMID 2320125. S2CID 4259327.
- "Spinal muscular atrophy". Genetics Home Reference. Retrieved 15 May 2019.
- Lefebvre S, Bürglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M (January 1995). "Identification and characterization of a spinal muscular atrophy-determining gene". Cell. 80 (1): 155–65. doi:10.1016/0092-8674(95)90460-3. PMID 7813012. S2CID 14291056.
- Passini MA, Bu J, Richards AM, Kinnecom C, Sardi SP, Stanek LM, Hua Y, Rigo F, Matson J, Hung G, Kaye EM, Shihabuddin LS, Krainer AR, Bennett CF, Cheng SH (March 2011). "Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy". Science Translational Medicine. 3 (72): 72ra18. doi:10.1126/scitranslmed.3001777. PMC 3140425. PMID 21368223.
- Wang CH, Finkel RS, Bertini ES, Schroth M, Simonds A, Wong B, Aloysius A, Morrison L, Main M, Crawford TO, Trela A (August 2007). "Consensus statement for standard of care in spinal muscular atrophy". Journal of Child Neurology. 22 (8): 1027–49. doi:10.1177/0883073807305788. PMID 17761659. S2CID 6478040.
- Jedrzejowska M, Milewski M, Zimowski J, Borkowska J, Kostera-Pruszczyk A, Sielska D, Jurek M, Hausmanowa-Petrusewicz I (2009). "Phenotype modifiers of spinal muscular atrophy: the number of SMN2 gene copies, deletion in the NAIP gene and probably gender influence the course of the disease". Acta Biochimica Polonica. 56 (1): 103–8. doi:10.18388/abp.2009_2521. PMID 19287802.
- Su YN, Hung CC, Lin SY, Chen FY, Chern JP, Tsai C, Chang TS, Yang CC, Li H, Ho HN, Lee CN (February 2011). Schrijver I (ed.). "Carrier screening for spinal muscular atrophy (SMA) in 107,611 pregnant women during the period 2005–2009: a prospective population-based cohort study". PLOS ONE. 6 (2): e17067. Bibcode:2011PLoSO...617067S. doi:10.1371/journal.pone.0017067. PMC 3045421. PMID 21364876.
- Sugarman EA, Nagan N, Zhu H, Akmaev VR, Zhou Z, Rohlfs EM, Flynn K, Hendrickson BC, Scholl T, Sirko-Osadsa DA, Allitto BA (January 2012). "Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: clinical laboratory analysis of >72,400 specimens". European Journal of Human Genetics. 20 (1): 27–32. doi:10.1038/ejhg.2011.134. PMC 3234503. PMID 21811307.
- Serra-Juhe C, Tizzano EF (December 2019). "Perspectives in genetic counseling for spinal muscular atrophy in the new therapeutic era: early pre-symptomatic intervention and test in minors". European Journal of Human Genetics. 27 (12): 1774–1782. doi:10.1038/s41431-019-0415-4. PMC 6871529. PMID 31053787.
- Glascock J, Sampson J, Haidet-Phillips A, Connolly A, Darras B, Day J, et al. (29 May 2018). "Treatment Algorithm for Infants Diagnosed with Spinal Muscular Atrophy through Newborn Screening". Journal of Neuromuscular Diseases. 5 (2): 145–158. doi:10.3233/JND-180304. PMC 6004919. PMID 29614695.
- Dangouloff T, Burghes A, Tizzano EF, Servais L (January 2020). "244th ENMC international workshop: Newborn screening in spinal muscular atrophy May 10-12, 2019, Hoofdorp, The Netherlands". Neuromuscular Disorders. 30 (1): 93–103. doi:10.1016/j.nmd.2019.11.002. PMID 31882184.
- Lopes JM (16 July 2018). "SMA Added to List of Recommended Screenings for Disease Given to..." SMA News Today. Retrieved 4 May 2020.
- Stephenson K (5 July 2018). "SMA Added to National List of Disorders to Screen for at Birth". Muscular Dystrophy Association. Retrieved 4 May 2020.
- "Recommended Uniform Screening Panel". Official web site of the U.S. Health Resources & Services Administration. 3 July 2017. Retrieved 4 May 2020.
- McCall S. "Newborn Screening for Spinal Muscular Atrophy". Cure SMA. Retrieved 4 May 2020.
- Kraszewski JN, Kay DM, Stevens CF, Koval C, Haser B, Ortiz V, et al. (June 2018). "Pilot study of population-based newborn screening for spinal muscular atrophy in New York state". Genetics in Medicine. 20 (6): 608–613. doi:10.1038/gim.2017.152. PMID 29758563.
- Chien YH, Chiang SC, Weng WC, Lee NC, Lin CJ, Hsieh WS, et al. (November 2017). "Presymptomatic Diagnosis of Spinal Muscular Atrophy Through Newborn Screening". The Journal of Pediatrics. 190: 124–129.e1. doi:10.1016/j.jpeds.2017.06.042. PMID 28711173. S2CID 20621772.
- Kariyawasam DS, Russell JS, Wiley V, Alexander IE, Farrar MA (March 2020). "The implementation of newborn screening for spinal muscular atrophy: the Australian experience". Genetics in Medicine. 22 (3): 557–565. doi:10.1038/s41436-019-0673-0. PMID 31607747. S2CID 204459317.
- Boemer F, Caberg JH, Dideberg V, Dardenne D, Bours V, Hiligsmann M, et al. (May 2019). "Newborn screening for SMA in Southern Belgium". Neuromuscular Disorders. 29 (5): 343–349. doi:10.1016/j.nmd.2019.02.003. PMID 31030938. S2CID 72332212.
- Vill K, Kölbel H, Schwartz O, Blaschek A, Olgemöller B, Harms E, et al. (31 October 2019). "One Year of Newborn Screening for SMA – Results of a German Pilot Project". Journal of Neuromuscular Diseases. 6 (4): 503–515. doi:10.3233/JND-190428. PMC 6918901. PMID 31594245.
- Ministerie van Volksgezondheid, Welzijn en Sport (23 July 2019). "Neonatal screening for spinal muscular atrophy – Advisory report – The Health Council of the Netherlands". www.healthcouncil.nl. Retrieved 4 May 2020.
- Lin Y, Lin CH, Yin X, Zhu L, Yang J, Shen Y, et al. (2019). "Newborn Screening for Spinal Muscular Atrophy in China Using DNA Mass Spectrometry". Frontiers in Genetics. 10: 1255. doi:10.3389/fgene.2019.01255. PMC 6928056. PMID 31921298.
- Shinohara M, Niba ET, Wijaya YO, Takayama I, Mitsuishi C, Kumasaka S, Kondo Y, Takatera A, Hokuto I, Morioka I, Ogiwara K (December 2019). "A Novel System for Spinal Muscular Atrophy Screening in Newborns: Japanese Pilot Study". International Journal of Neonatal Screening. 5 (4): 41. doi:10.3390/ijns5040041. PMC 7510215. PMID 33072999.
- "Carrier Screening in the Age of Genomic Medicine – ACOG". www.acog.org. Retrieved 24 February 2017.
- Nilay, M, Moirangthem, A, Saxena, D, Mandal, K, Phadke, SR (October 2020). "Carrier frequency of SMN1 related spinal muscular atrophy in north Indian population: The need for population based screening program". American Journal of Medical Genetics Part A. 185 (1): 274–277. doi:10.1002/ajmg.a.61918. PMID 33051992. S2CID 222353383.
- Prior TW (November 2008). "Carrier screening for spinal muscular atrophy". Genetics in Medicine. 10 (11): 840–2. doi:10.1097/GIM.0b013e318188d069. PMC 3110347. PMID 18941424.
- Ar Rochmah M, Awano H, Awaya T, Harahap NI, Morisada N, Bouike Y, Saito T, Kubo Y, Saito K, Lai PS, Morioka I, Iijima K, Nishio H, Shinohara M (November 2017). "Spinal muscular atrophy carriers with two SMN1 copies". Brain & Development. 39 (10): 851–860. doi:10.1016/j.braindev.2017.06.002. PMID 28676237. S2CID 26504674.
- "Spinraza- nusinersen injection, solution". DailyMed. 30 June 2020. Retrieved 8 August 2020.
- Grant C (27 December 2016). "Surprise Drug Approval Is Holiday Gift for Biogen". The Wall Street Journal. ISSN 0099-9660. Retrieved 27 December 2016.
- Finkel RS, Mercuri E, Darras BT, Connolly AM, Kuntz NL, Kirschner J, et al. (November 2017). "Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy". New England Journal of Medicine. 377 (18): 1723–32. doi:10.1056/NEJMoa1702752. PMID 29091570. S2CID 4771819.
- Wadman, Renske I.; van der Pol, W. Ludo; Bosboom, Wendy Mj; Asselman, Fay-Lynn; van den Berg, Leonard H.; Iannaccone, Susan T.; Vrancken, Alexander Fje (1 June 2020). "Drug treatment for spinal muscular atrophy types II and III". The Cochrane Database of Systematic Reviews. 1: CD006282. doi:10.1002/14651858.CD006282.pub5. ISSN 1469-493X. PMC 6995983. PMID 32006461.
- "Spinraza (nusinersen) Injection". U.S. Food and Drug Administration (FDA). 18 January 2017. Retrieved 8 August 2020.
- "Spinraza EPAR". European Medicines Agency (EMA). Retrieved 8 August 2020.
- "Spinraza (Nusinersen) Approved in the European Union as First Treatment for Spinal Muscular Atrophy". Agence France-Presse (AFP). 1 June 2017. Retrieved 1 June 2017.
- "Zolgensma 2 x 1013 vector genomes/mL solution for infusion". www.medicines.org.uk. Retrieved 8 August 2020.
- "Zolgensma- onasemnogene abeparvovec-xioi kit". DailyMed. 24 May 2019. Retrieved 8 August 2020.
- "FDA approves innovative gene therapy to treat pediatric patients with spinal muscular atrophy, a rare disease and leading genetic cause of infant mortality". U.S. Food and Drug Administration (FDA) (Press release). 24 May 2019. Retrieved 27 May 2019.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - "Zolgensma". U.S. Food and Drug Administration (FDA). 24 May 2019. Retrieved 8 August 2020.
- "Zolgensma EPAR". European Medicines Agency (EMA). 24 March 2020. Retrieved 8 August 2020.
- "Novartis receives approval from Japanese Ministry of Health, Labour and Welfare for Zolgensma the only gene therapy for patients with spinal muscular atrophy (SMA)". Novartis (Press release). Retrieved 8 August 2020.
- "FDA Approves Oral Treatment for Spinal Muscular Atrophy". U.S. Food and Drug Administration (FDA) (Press release). 7 August 2020. Retrieved 7 August 2020.
- "Evrysdi (risdiplam) for oral solution" (PDF). Genentech. Retrieved 8 August 2020.
- Maria Joao Almeida (8 September 2016). "RG7916". BioNews Services. Retrieved 8 October 2017.
- Zhao X, Feng Z, Ling KK, Mollin A, Sheedy J, Yeh S, et al. (May 2016). "Pharmacokinetics, pharmacodynamics, and efficacy of a small-molecule SMN2 splicing modifier in mouse models of spinal muscular atrophy". Human Molecular Genetics. 25 (10): 1885–1899. doi:10.1093/hmg/ddw062. PMC 5062580. PMID 26931466.
- Bodamer O (November 2017). "Spinal Muscular Atrophy". uptodate.com. Retrieved 1 December 2017.
- Bach JR, Niranjan V, Weaver B (April 2000). "Spinal muscular atrophy type 1: A noninvasive respiratory management approach". Chest. 117 (4): 1100–5. doi:10.1378/chest.117.4.1100. PMID 10767247.
- Bach JR, Saltstein K, Sinquee D, Weaver B, Komaroff E (May 2007). "Long-term survival in Werdnig-Hoffmann disease". American Journal of Physical Medicine & Rehabilitation. 86 (5): 339–45 quiz 346–8, 379. doi:10.1097/PHM.0b013e31804a8505. PMID 17449977. S2CID 9942245.
- Messina S, Pane M, De Rose P, Vasta I, Sorleti D, Aloysius A, Sciarra F, Mangiola F, Kinali M, Bertini E, Mercuri E (May 2008). "Feeding problems and malnutrition in spinal muscular atrophy type II". Neuromuscular Disorders. 18 (5): 389–93. doi:10.1016/j.nmd.2008.02.008. PMID 18420410. S2CID 23302291.
- Chen YS, Shih HH, Chen TH, Kuo CH, Jong YJ (March 2012). "Prevalence and risk factors for feeding and swallowing difficulties in spinal muscular atrophy types II and III". The Journal of Pediatrics. 160 (3): 447–451.e1. doi:10.1016/j.jpeds.2011.08.016. PMID 21924737.
- Tilton AH, Miller MD, Khoshoo V (June 1998). "Nutrition and swallowing in pediatric neuromuscular patients". Seminars in Pediatric Neurology. 5 (2): 106–15. doi:10.1016/S1071-9091(98)80026-0. PMID 9661244.
- Tein I, Sloane AE, Donner EJ, Lehotay DC, Millington DS, Kelley RI (January 1995). "Fatty acid oxidation abnormalities in childhood-onset spinal muscular atrophy: primary or secondary defect(s)?". Pediatric Neurology. 12 (1): 21–30. doi:10.1016/0887-8994(94)00100-G. PMID 7748356.
- Crawford TO, Sladky JT, Hurko O, Besner-Johnston A, Kelley RI (March 1999). "Abnormal fatty acid metabolism in childhood spinal muscular atrophy". Annals of Neurology. 45 (3): 337–43. doi:10.1002/1531-8249(199903)45:3<337::AID-ANA9>3.0.CO;2-U. PMID 10072048. S2CID 23808651.
- Leighton S (2003). "Nutrition issues associated with spinal muscular atrophy". Nutrition & Dietetics. 60 (2): 92–96.
- Apkon S (Summer 2017). "SMA CARE SERIES – Musculoskeletal System" (PDF). www.curesma.org. Archived from the original (PDF) on 19 February 2018. Retrieved 7 December 2017.
- Rudnik-Schöneborn S, Heller R, Berg C, Betzler C, Grimm T, Eggermann T, Eggermann K, Wirth R, Wirth B, Zerres K (October 2008). "Congenital heart disease is a feature of severe infantile spinal muscular atrophy". Journal of Medical Genetics. 45 (10): 635–8. doi:10.1136/jmg.2008.057950. PMID 18662980. S2CID 7170069.
- Heier CR, Satta R, Lutz C, DiDonato CJ (October 2010). "Arrhythmia and cardiac defects are a feature of spinal muscular atrophy model mice". Human Molecular Genetics. 19 (20): 3906–18. doi:10.1093/hmg/ddq330. PMC 2947406. PMID 20693262.
- Shababi M, Habibi J, Yang HT, Vale SM, Sewell WA, Lorson CL (October 2010). "Cardiac defects contribute to the pathology of spinal muscular atrophy models". Human Molecular Genetics. 19 (20): 4059–71. doi:10.1093/hmg/ddq329. PMID 20696672.
- Bevan AK, Hutchinson KR, Foust KD, Braun L, McGovern VL, Schmelzer L, Ward JG, Petruska JC, Lucchesi PA, Burghes AH, Kaspar BK (October 2010). "Early heart failure in the SMNDelta7 model of spinal muscular atrophy and correction by postnatal scAAV9-SMN delivery". Human Molecular Genetics. 19 (20): 3895–905. doi:10.1093/hmg/ddq300. PMC 2947399. PMID 20639395.
- von Gontard A, Zerres K, Backes M, Laufersweiler-Plass C, Wendland C, Melchers P, Lehmkuhl G, Rudnik-Schöneborn S (February 2002). "Intelligence and cognitive function in children and adolescents with spinal muscular atrophy". Neuromuscular Disorders. 12 (2): 130–6. doi:10.1016/S0960-8966(01)00274-7. PMID 11738354. S2CID 46694209.
- Billard C, Gillet P, Signoret JL, Uicaut E, Bertrand P, Fardeau M, Barthez-Carpentier MA, Santini JJ (1992). "Cognitive functions in Duchenne muscular dystrophy: a reappraisal and comparison with spinal muscular atrophy". Neuromuscular Disorders. 2 (5–6): 371–8. doi:10.1016/S0960-8966(06)80008-8. PMID 1300185. S2CID 22211725.
- Laufersweiler-Plass C, Rudnik-Schöneborn S, Zerres K, Backes M, Lehmkuhl G, von Gontard A (January 2003). "Behavioural problems in children and adolescents with spinal muscular atrophy and their siblings". Developmental Medicine and Child Neurology. 45 (1): 44–9. doi:10.1017/S0012162203000082. PMID 12549754.
- de Oliveira CM, Araújo AP (January 2011). "Self-reported quality of life has no correlation with functional status in children and adolescents with spinal muscular atrophy". European Journal of Paediatric Neurology. 15 (1): 36–9. doi:10.1016/j.ejpn.2010.07.003. PMID 20800519.
- Darras B, Finkel R (2017). Spinal Muscular Atrophy. United Kingdom, United States: Elsevier. p. 417. ISBN 978-0-12-803685-3.
- Yuan N, Wang CH, Trela A, Albanese CT (June 2007). "Laparoscopic Nissen fundoplication during gastrostomy tube placement and noninvasive ventilation may improve survival in type I and severe type II spinal muscular atrophy". Journal of Child Neurology. 22 (6): 727–31. doi:10.1177/0883073807304009. PMID 17641258. S2CID 38799022.
- Bach JR (May 2007). "Medical considerations of long-term survival of Werdnig-Hoffmann disease". American Journal of Physical Medicine & Rehabilitation. 86 (5): 349–55. doi:10.1097/PHM.0b013e31804b1d66. PMID 17449979. S2CID 39989993.
- Oskoui M, Levy G, Garland CJ, Gray JM, O'Hagen J, De Vivo DC, Kaufmann P (November 2007). "The changing natural history of spinal muscular atrophy type 1". Neurology. 69 (20): 1931–6. doi:10.1212/01.wnl.0000290830.40544.b9. PMID 17998484. S2CID 7528894.
- d'Ydewalle C, Sumner CJ (April 2015). "Spinal Muscular Atrophy Therapeutics: Where do we Stand?". Neurotherapeutics. 12 (2): 303–16. doi:10.1007/s13311-015-0337-y. PMC 4404440. PMID 25631888.
- "$2.1m Novartis gene therapy to become world's most expensive drug". The Guardian. Reuters. 25 May 2019. ISSN 0261-3077.
- "Novartis Releases Update on LMI070 (Branaplam) Clinical Trial". CureSMA. Archived from the original on 25 November 2017. Retrieved 7 October 2017.
- Kletzl, Heidemarie; Marquet, Anne; Günther, Andreas; Tang, Wakana; Heuberger, Jules; Groeneveld, Geert Jan; Birkhoff, Willem; Mercuri, Eugenio; Lochmüller, Hanns; Wood, Claire; Fischer, Dirk; Gerlach, Irene; Heinig, Katja; Bugawan, Teodorica; Dziadek, Sebastian; Kinch, Russell; Czech, Christian; Khwaja, Omar (2019). "The oral splicing modifier RG7800 increases full length survival of motor neuron 2 mRNA and survival of motor neuron protein: Results from trials in healthy adults and patients with spinal muscular atrophy". Neuromuscular Disorders. Elsevier BV. 29 (1): 21–29. doi:10.1016/j.nmd.2018.10.001. ISSN 0960-8966. PMID 30553700. S2CID 54315649.
- Zhang ML, Lorson CL, Androphy EJ, Zhou J (October 2001). "An in vivo reporter system for measuring increased inclusion of exon 7 in SMN2 mRNA: potential therapy of SMA". Gene Therapy. 8 (20): 1532–8. doi:10.1038/sj.gt.3301550. PMID 11704813.
- Andreassi C, Jarecki J, Zhou J, Coovert DD, Monani UR, Chen X, Whitney M, Pollok B, Zhang M, Androphy E, Burghes AH (November 2001). "Aclarubicin treatment restores SMN levels to cells derived from type I spinal muscular atrophy patients". Human Molecular Genetics. 10 (24): 2841–9. doi:10.1093/hmg/10.24.2841. PMID 11734549.
- Zhou H, Meng J, Marrosu E, Janghra N, Morgan J, Muntoni F (November 2015). "Repeated low doses of morpholino antisense oligomer: an intermediate mouse model of spinal muscular atrophy to explore the window of therapeutic response". Human Molecular Genetics. 24 (22): 6265–77. doi:10.1093/hmg/ddv329. PMC 4614699. PMID 26264577.
- Hammond SM, Hazell G, Shabanpoor F, Saleh AF, Bowerman M, Sleigh JN, Meijboom KE, Zhou H, Muntoni F, Talbot K, Gait MJ, Wood MJ (September 2016). "Systemic peptide-mediated oligonucleotide therapy improves long-term survival in spinal muscular atrophy". Proceedings of the National Academy of Sciences of the United States of America. 113 (39): 10962–7. doi:10.1073/pnas.1605731113. PMC 5047168. PMID 27621445.
- Angelozzi C, Borgo F, Tiziano FD, Martella A, Neri G, Brahe C (January 2008). "Salbutamol increases SMN mRNA and protein levels in spinal muscular atrophy cells". Journal of Medical Genetics. 45 (1): 29–31. doi:10.1136/jmg.2007.051177. PMID 17932121. S2CID 29911453.
- Pane M, Staccioli S, Messina S, D'Amico A, Pelliccioni M, Mazzone ES, Cuttini M, Alfieri P, Battini R, Main M, Muntoni F, Bertini E, Villanova M, Mercuri E (July 2008). "Daily salbutamol in young patients with SMA type II". Neuromuscular Disorders. 18 (7): 536–40. doi:10.1016/j.nmd.2008.05.004. PMID 18579379. S2CID 34334434.
- Tiziano FD, Lomastro R, Pinto AM, Messina S, D'Amico A, Fiori S, Angelozzi C, Pane M, Mercuri E, Bertini E, Neri G, Brahe C (December 2010). "Salbutamol increases survival motor neuron (SMN) transcript levels in leucocytes of spinal muscular atrophy (SMA) patients: relevance for clinical trial design" (PDF). Journal of Medical Genetics. 47 (12): 856–8. doi:10.1136/jmg.2010.080366. PMID 20837492. S2CID 21825049.
- Morandi L, Abiusi E, Pasanisi MB, Lomastro R, Fiori S, Di Pietro L, Angelini C, Sorarù G, Gaiani A, Mongini T, Vercelli L (2013). "P.6.4 Salbutamol tolerability and efficacy in adult type III SMA patients: Results of a multicentric, molecular and clinical, double-blind, placebo-controlled study". Neuromuscular Disorders. 23 (9–10): 771. doi:10.1016/j.nmd.2013.06.475. S2CID 54398218.
- Chang JG, Hsieh-Li HM, Jong YJ, Wang NM, Tsai CH, Li H (August 2001). "Treatment of spinal muscular atrophy by sodium butyrate". Proceedings of the National Academy of Sciences of the United States of America. 98 (17): 9808–13. Bibcode:2001PNAS...98.9808C. doi:10.1073/pnas.171105098. PMC 55534. PMID 11504946.
- Andreassi C, Angelozzi C, Tiziano FD, Vitali T, De Vincenzi E, Boninsegna A, Villanova M, Bertini E, Pini A, Neri G, Brahe C (January 2004). "Phenylbutyrate increases SMN expression in vitro: relevance for treatment of spinal muscular atrophy". European Journal of Human Genetics. 12 (1): 59–65. doi:10.1038/sj.ejhg.5201102. PMID 14560316.
- Brahe C, Vitali T, Tiziano FD, Angelozzi C, Pinto AM, Borgo F, Moscato U, Bertini E, Mercuri E, Neri G (February 2005). "Phenylbutyrate increases SMN gene expression in spinal muscular atrophy patients". European Journal of Human Genetics. 13 (2): 256–9. doi:10.1038/sj.ejhg.5201320. PMID 15523494.
- Mercuri E, Bertini E, Messina S, Solari A, D'Amico A, Angelozzi C, Battini R, Berardinelli A, Boffi P, Bruno C, Cini C, Colitto F, Kinali M, Minetti C, Mongini T, Morandi L, Neri G, Orcesi S, Pane M, Pelliccioni M, Pini A, Tiziano FD, Villanova M, Vita G, Brahe C (January 2007). "Randomized, double-blind, placebo-controlled trial of phenylbutyrate in spinal muscular atrophy". Neurology. 68 (1): 51–5. doi:10.1212/01.wnl.0000249142.82285.d6. PMID 17082463. S2CID 30429093.
- Clinical trial number NCT00528268 for "Study to Evaluate Sodium Phenylbutyrate in Pre-symptomatic Infants With Spinal Muscular Atrophy (STOPSMA)" at ClinicalTrials.gov
- Brichta L, Hofmann Y, Hahnen E, Siebzehnrubl FA, Raschke H, Blumcke I, Eyupoglu IY, Wirth B (October 2003). "Valproic acid increases the SMN2 protein level: a well-known drug as a potential therapy for spinal muscular atrophy". Human Molecular Genetics. 12 (19): 2481–9. doi:10.1093/hmg/ddg256. PMID 12915451.
- Tsai LK, Tsai MS, Ting CH, Li H (November 2008). "Multiple therapeutic effects of valproic acid in spinal muscular atrophy model mice". Journal of Molecular Medicine. 86 (11): 1243–54. doi:10.1007/s00109-008-0388-1. PMID 18649067. S2CID 24565272.
- Swoboda KJ, Scott CB, Crawford TO, Simard LR, Reyna SP, Krosschell KJ, Acsadi G, Elsheik B, Schroth MK, D'Anjou G, LaSalle B, Prior TW, Sorenson SL, Maczulski JA, Bromberg MB, Chan GM, Kissel JT, et al. (Project Cure Spinal Muscular Atrophy Investigators Network) (August 2010). Boutron I (ed.). "SMA CARNI-VAL trial part I: double-blind, randomized, placebo-controlled trial of L-carnitine and valproic acid in spinal muscular atrophy". PLOS ONE. 5 (8): e12140. Bibcode:2010PLoSO...512140S. doi:10.1371/journal.pone.0012140. PMC 2924376. PMID 20808854.
- Kissel JT, Scott CB, Reyna SP, Crawford TO, Simard LR, Krosschell KJ, Acsadi G, Elsheik B, Schroth MK, D'Anjou G, LaSalle B, Prior TW, Sorenson S, Maczulski JA, Bromberg MB, Chan GM, Swoboda KJ, et al. (Project Cure Spinal Muscular Atrophy Investigators' Network) (2011). "SMA CARNIVAL TRIAL PART II: a prospective, single-armed trial of L-carnitine and valproic acid in ambulatory children with spinal muscular atrophy". PLOS ONE. 6 (7): e21296. Bibcode:2011PLoSO...621296K. doi:10.1371/journal.pone.0021296. PMC 3130730. PMID 21754985.
- Darbar IA, Plaggert PG, Resende MB, Zanoteli E, Reed UC (March 2011). "Evaluation of muscle strength and motor abilities in children with type II and III spinal muscle atrophy treated with valproic acid". BMC Neurology. 11: 36. doi:10.1186/1471-2377-11-36. PMC 3078847. PMID 21435220.
- Garbes L, Heesen L, Hölker I, Bauer T, Schreml J, Zimmermann K, Thoenes M, Walter M, Dimos J, Peitz M, Brüstle O, Heller R, Wirth B (January 2013). "VPA response in SMA is suppressed by the fatty acid translocase CD36". Human Molecular Genetics. 22 (2): 398–407. doi:10.1093/hmg/dds437. PMID 23077215.
- Rak K, Lechner BD, Schneider C, Drexl H, Sendtner M, Jablonka S (December 2009). "Valproic acid blocks excitability in SMA type I mouse motor neurons". Neurobiology of Disease. 36 (3): 477–87. doi:10.1016/j.nbd.2009.08.014. PMID 19733665. S2CID 34657615.
- Elshafay A, Hieu TH, Doheim MF, Kassem MA, ELdoadoa MF, Holloway SK, Abo-Elghar H, Hirayama K, Huy NT (March 2019). "Efficacy and Safety of Valproic Acid for Spinal Muscular Atrophy: A Systematic Review and Meta-Analysis". CNS Drugs. 33 (3): 239–250. doi:10.1007/s40263-019-00606-6. PMID 30796634. S2CID 73495750.
- Grzeschik SM, Ganta M, Prior TW, Heavlin WD, Wang CH (August 2005). "Hydroxyurea enhances SMN2 gene expression in spinal muscular atrophy cells". Annals of Neurology. 58 (2): 194–202. doi:10.1002/ana.20548. PMID 16049920. S2CID 19509393.
- Chen TH, Chang JG, Yang YH, Mai HH, Liang WC, Wu YC, Wang HY, Huang YB, Wu SM, Chen YC, Yang SN, Jong YJ (December 2010). "Randomized, double-blind, placebo-controlled trial of hydroxyurea in spinal muscular atrophy". Neurology. 75 (24): 2190–7. doi:10.1212/WNL.0b013e3182020332. PMID 21172842. S2CID 25858890.
- Evans MC, Cherry JJ, Androphy EJ (October 2011). "Differential regulation of the SMN2 gene by individual HDAC proteins". Biochemical and Biophysical Research Communications. 414 (1): 25–30. doi:10.1016/j.bbrc.2011.09.011. PMC 6538936. PMID 21925145.
- Riessland M, Brichta L, Hahnen E, Wirth B (August 2006). "The benzamide M344, a novel histone deacetylase inhibitor, significantly increases SMN2 RNA/protein levels in spinal muscular atrophy cells". Human Genetics. 120 (1): 101–10. doi:10.1007/s00439-006-0186-1. PMID 16724231. S2CID 24804136.
- Garbes L, Riessland M, Hölker I, Heller R, Hauke J, Tränkle C, Coras R, Blümcke I, Hahnen E, Wirth B (October 2009). "LBH589 induces up to 10-fold SMN protein levels by several independent mechanisms and is effective even in cells from SMA patients non-responsive to valproate". Human Molecular Genetics. 18 (19): 3645–58. doi:10.1093/hmg/ddp313. PMID 19584083.
- Narver HL, Kong L, Burnett BG, Choe DW, Bosch-Marcé M, Taye AA, Eckhaus MA, Sumner CJ (October 2008). "Sustained improvement of spinal muscular atrophy mice treated with trichostatin A plus nutrition". Annals of Neurology. 64 (4): 465–70. doi:10.1002/ana.21449. PMID 18661558. S2CID 5595968.
- Avila AM, Burnett BG, Taye AA, Gabanella F, Knight MA, Hartenstein P, Cizman Z, Di Prospero NA, Pellizzoni L, Fischbeck KH, Sumner CJ (March 2007). "Trichostatin A increases SMN expression and survival in a mouse model of spinal muscular atrophy". The Journal of Clinical Investigation. 117 (3): 659–71. doi:10.1172/JCI29562. PMC 1797603. PMID 17318264.
- Riessland M, Ackermann B, Förster A, Jakubik M, Hauke J, Garbes L, Fritzsche I, Mende Y, Blumcke I, Hahnen E, Wirth B (April 2010). "SAHA ameliorates the SMA phenotype in two mouse models for spinal muscular atrophy". Human Molecular Genetics. 19 (8): 1492–506. doi:10.1093/hmg/ddq023. PMID 20097677.
- Farooq F, Molina FA, Hadwen J, MacKenzie D, Witherspoon L, Osmond M, Holcik M, MacKenzie A (August 2011). "Prolactin increases SMN expression and survival in a mouse model of severe spinal muscular atrophy via the STAT5 pathway". The Journal of Clinical Investigation. 121 (8): 3042–50. doi:10.1172/JCI46276. PMC 3148738. PMID 21785216.
- Sakla MS, Lorson CL (January 2008). "Induction of full-length survival motor neuron by polyphenol botanical compounds". Human Genetics. 122 (6): 635–43. doi:10.1007/s00439-007-0441-0. PMID 17962980. S2CID 12460406.
- Dayangaç-Erden D, Bora G, Ayhan P, Kocaefe C, Dalkara S, Yelekçi K, Demir AS, Erdem-Yurter H (March 2009). "Histone deacetylase inhibition activity and molecular docking of (e )-resveratrol: its therapeutic potential in spinal muscular atrophy". Chemical Biology & Drug Design. 73 (3): 355–64. CiteSeerX 10.1.1.515.8424. doi:10.1111/j.1747-0285.2009.00781.x. PMID 19207472. S2CID 764215.
- Farooq F, Abadía-Molina F, MacKenzie D, Hadwen J, Shamim F, O'Reilly S, Holcik M, MacKenzie A (September 2013). "Celecoxib increases SMN and survival in a severe spinal muscular atrophy mouse model via p38 pathway activation". Human Molecular Genetics. 22 (17): 3415–24. doi:10.1093/hmg/ddt191. PMID 23656793.
- Burnett BG, Muñoz E, Tandon A, Kwon DY, Sumner CJ, Fischbeck KH (March 2009). "Regulation of SMN protein stability". Molecular and Cellular Biology. 29 (5): 1107–15. doi:10.1128/MCB.01262-08. PMC 2643817. PMID 19103745.
- Mattis VB, Rai R, Wang J, Chang CW, Coady T, Lorson CL (November 2006). "Novel aminoglycosides increase SMN levels in spinal muscular atrophy fibroblasts". Human Genetics. 120 (4): 589–601. doi:10.1007/s00439-006-0245-7. PMID 16951947. S2CID 28834037.
- Mattis VB, Fosso MY, Chang CW, Lorson CL (November 2009). "Subcutaneous administration of TC007 reduces disease severity in an animal model of SMA". BMC Neuroscience. 10: 142. doi:10.1186/1471-2202-10-142. PMC 2789732. PMID 19948047.
- Lunn MR, Root DE, Martino AM, Flaherty SP, Kelley BP, Coovert DD, Burghes AH, Man NT, Morris GE, Zhou J, Androphy EJ, Sumner CJ, Stockwell BR (November 2004). "Indoprofen upregulates the survival motor neuron protein through a cyclooxygenase-independent mechanism". Chemistry & Biology. 11 (11): 1489–93. doi:10.1016/j.chembiol.2004.08.024. PMC 3160629. PMID 15555999.
- Taylor NP (1 June 2018). "Roche scraps €120M SMA drug after hitting 'many difficulties'". www.fiercebiotech.com. Retrieved 8 June 2018.
- Takeuchi Y, Miyanomae Y, Komatsu H, Oomizono Y, Nishimura A, Okano S, Nishiki T, Sawada T (July 1994). "Efficacy of thyrotropin-releasing hormone in the treatment of spinal muscular atrophy". Journal of Child Neurology. 9 (3): 287–9. doi:10.1177/088307389400900313. PMID 7930408. S2CID 41678161.
- Tzeng AC, Cheng J, Fryczynski H, Niranjan V, Stitik T, Sial A, Takeuchi Y, Foye P, DePrince M, Bach JR (2000). "A study of thyrotropin-releasing hormone for the treatment of spinal muscular atrophy: a preliminary report". American Journal of Physical Medicine & Rehabilitation. 79 (5): 435–40. doi:10.1097/00002060-200009000-00005. PMID 10994885. S2CID 20416253.
- Kato Z, Okuda M, Okumura Y, Arai T, Teramoto T, Nishimura M, Kaneko H, Kondo N (August 2009). "Oral administration of the thyrotropin-releasing hormone (TRH) analogue, taltireline hydrate, in spinal muscular atrophy". Journal of Child Neurology. 24 (8): 1010–2. doi:10.1177/0883073809333535. PMID 19666885. S2CID 29321906.
- Wadman RI, Bosboom WM, van den Berg LH, Wokke LH, Iannaccone ST, Vrancken AF, et al. (The Cochrane Collaboration) (7 December 2011). Wadman RI (ed.). "Drug treatment for spinal muscular atrophy type I". Cochrane Database of Systematic Reviews. John Wiley & Sons, Ltd (12): CD006281. doi:10.1002/14651858.cd006281.pub3. PMID 22161399.
- Haddad H, Cifuentes-Diaz C, Miroglio A, Roblot N, Joshi V, Melki J (October 2003). "Riluzole attenuates spinal muscular atrophy disease progression in a mouse model". Muscle & Nerve. 28 (4): 432–7. doi:10.1002/mus.10455. PMID 14506714. S2CID 10300057.
- Dimitriadi M, Kye MJ, Kalloo G, Yersak JM, Sahin M, Hart AC (April 2013). "The neuroprotective drug riluzole acts via small conductance Ca2+-activated K+ channels to ameliorate defects in spinal muscular atrophy models". The Journal of Neuroscience. 33 (15): 6557–62. doi:10.1523/JNEUROSCI.1536-12.2013. PMC 3652322. PMID 23575853.
- Clinical trial number NCT00774423 for "Study to Evaluate the Efficacy of Riluzole in Children and Young Adults With Spinal Muscular Atrophy (SMA)" at ClinicalTrials.gov
- "Riluzole: premiers résultats décevants" (in French). AFM Téléthon. 22 September 2010. Archived from the original on 8 December 2017. Retrieved 16 March 2017.
- Nizzardo M, Nardini M, Ronchi D, Salani S, Donadoni C, Fortunato F, Colciago G, Falcone M, Simone C, Riboldi G, Govoni A, Bresolin N, Comi GP, Corti S (June 2011). "Beta-lactam antibiotic offers neuroprotection in a spinal muscular atrophy model by multiple mechanisms" (PDF). Experimental Neurology. 229 (2): 214–25. doi:10.1016/j.expneurol.2011.01.017. hdl:2434/425410. PMID 21295027. S2CID 47567316.
- Hedlund E (September 2011). "The protective effects of β-lactam antibiotics in motor neuron disorders". Experimental Neurology. 231 (1): 14–8. doi:10.1016/j.expneurol.2011.06.002. PMID 21693120. S2CID 26353910.
- Rose FF, Mattis VB, Rindt H, Lorson CL (March 2009). "Delivery of recombinant follistatin lessens disease severity in a mouse model of spinal muscular atrophy". Human Molecular Genetics. 18 (6): 997–1005. doi:10.1093/hmg/ddn426. PMC 2649020. PMID 19074460.
- "CK-2127107".
- "Scholar Rock Announces Positive 12-Month Top-Line Results From the TOPAZ Phase 2 Clinical Trial Evaluating Apitegromab in Patients With Type 2 and Type 3 Spinal Muscular Atrophy (SMA)". www.businesswire.com. 6 April 2021. Retrieved 13 May 2021.
- PhD, Patricia Inacio. "Pediatric Phase 2/3 Trial to Test Anti-myostatin Antibody with Evrysdi". Retrieved 23 January 2022.
- Committee for Advanced Therapies and CAT Scientific Secretariat (August 2010). "Use of unregulated stem-cell based medicinal products". Lancet. 376 (9740): 514. doi:10.1016/S0140-6736(10)61249-4. PMID 20709228. S2CID 6906599.
- European Medicines Agency (16 April 2010). "Concerns over unregulated medicinal products containing stem cells" (PDF). European Medicines Agency.
- Carrozzi M, Amaddeo A, Biondi A, Zanus C, Monti F, Alessandro V (November 2012). "Stem cells in severe infantile spinal muscular atrophy (SMA1)". Neuromuscular Disorders. 22 (11): 1032–4. doi:10.1016/j.nmd.2012.09.005. PMID 23046997. S2CID 42093152.
- Mercuri E, Bertini E (December 2012). "Stem cells in severe infantile spinal muscular atrophy". Neuromuscular Disorders. 22 (12): 1105. doi:10.1016/j.nmd.2012.11.001. PMID 23206850. S2CID 43858783.
- "National registries for DMD, SMA and DM". Archived from the original on 22 January 2011.
Further reading
- Parano E, Pavone L, Falsaperla R, Trifiletti R, Wang C (August 1996). "Molecular basis of phenotypic heterogeneity in siblings with spinal muscular atrophy". Annals of Neurology. 40 (2): 247–51. doi:10.1002/ana.410400219. PMID 8773609. S2CID 42514712.
- Wang CH, Finkel RS, Bertini ES, Schroth M, Simonds A, Wong B, Aloysius A, Morrison L, Main M, Crawford TO, Trela A (August 2007). "Consensus statement for standard of care in spinal muscular atrophy". Journal of Child Neurology. 22 (8): 1027–49. doi:10.1177/0883073807305788. PMID 17761659. S2CID 6478040.
External links
- Spinal muscular atrophy at Curlie
- SMA at NINDS
- SMArt Moves. Cure SMA. Retrieved 3 December 2021.