Lysosomal storage disease
Lysosomal storage diseases (LSDs; /ˌlaɪsəˈsoʊməl/) are a group of over 70 rare inherited metabolic disorders that result from defects in lysosomal function.[1][2] Lysosomes are sacs of enzymes within cells that digest large molecules and pass the fragments on to other parts of the cell for recycling. This process requires several critical enzymes. If one of these enzymes is defective due to a mutation, the large molecules accumulate within the cell, eventually killing it.[3]
| Lysosomal storage disease | |
|---|---|
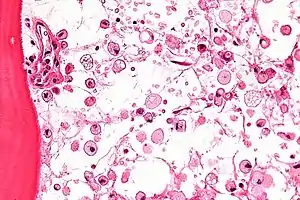 | |
| Micrograph of Gaucher disease, with cells that have the characteristic crumpled tissue paper-like cytoplasm. H&E stain. | |
| Specialty | Endocrinology |
Lysosomal storage disorders are caused by lysosomal dysfunction usually as a consequence of deficiency of a single enzyme required for the metabolism of lipids, glycoproteins (sugar-containing proteins), or so-called mucopolysaccharides. Individually, lysosomal storage diseases occur with incidences of less than 1:100,000; however, as a group, the incidence is about 1:5,000 – 1:10,000.[4][5] Most of these disorders are autosomal recessively inherited such as Niemann–Pick disease, type C, but a few are X-linked recessively inherited, such as Fabry disease and Hunter syndrome (MPS II).
The lysosome is commonly referred to as the cell's recycling center because it processes unwanted material into substances that the cell can use. Lysosomes break down this unwanted matter by enzymes, highly specialized proteins essential for survival. Lysosomal disorders are usually triggered when a particular enzyme exists in too small an amount or is missing altogether. When this happens, substances accumulate in the cell. In other words, when the lysosome does not function normally, excess products destined for breakdown and recycling are stored in the cell.
Like other genetic disorders, individuals inherit lysosomal storage diseases from their parents. Although each disorder results from different gene mutations that translate into a deficiency in enzyme activity, they all share a common biochemical characteristic – all lysosomal disorders originate from an abnormal accumulation of substances inside the lysosome.
Lysosomal storage diseases affect mostly children and they often die at a young age, many within a few months or years of birth.
Classification
Standard classification
The lysosomal storage diseases are generally classified by the nature of the primary stored material involved, and can be broadly broken into the following: (ICD-10 codes are provided where available)
- (E75) Lipid storage disorders
- Sphingolipidoses, including Gaucher's and Niemann–Pick diseases (E75.0-E75.1)
- Gangliosidosis (including Tay–Sachs disease (E75.2)
- Leukodystrophies
- (E76.0) Mucopolysaccharidoses, including Hunter syndrome and Hurler disease
- (E77) Glycoprotein storage disorders
- (E77.0-E77.1) Mucolipidoses
Also, glycogen storage disease type II (Pompe disease) is a defect in lysosomal metabolism as well,[6] although it is otherwise classified into E74.0 in ICD-10. Cystinosis is an lysosomal storage disease characterized by the abnormal accumulation of the amino acid cystine.
By type of defect protein
Alternatively to the protein targets, lysosomal storage diseases may be classified by the type of protein that is deficient and is causing buildup.
| Type of defect protein | Disease examples | Deficient protein |
|---|---|---|
| Lysosomal enzymes primarily | Tay–Sachs disease, I-cell disease,[7] Sphingolipidoses (e.g., Krabbe disease, gangliosidosis: Gaucher, Niemann–Pick disease and glycolipids: Metachromatic leukodystrophy), Lysosomal acid lipase deficiency | Various |
| Posttranslational modification of enzymes | Multiple sulfatase deficiency | Multiple sulfatases |
| Membrane transport proteins | Mucolipidosis type II and IIIA | N-acetylglucosamine-1-phosphate transferase |
| Enzyme protecting proteins | Galactosialidosis | Cathepsin A |
| Soluble nonenzymatic proteins | GM2-AP deficiency, variant AB, Niemann–Pick disease, type C2 | GM2-AP, NPC2 |
| Transmembrane proteins | SAP deficiency | Sphingolipid activator proteins |
| Niemann–Pick disease, type C1 | NPC1 | |
| Salla disease | Sialin | |
| Unless else specified in boxes, then the applicable reference is:[8] | ||
Lysosomal storage disorders
Lysosomal storage diseases include:
Sphingolipidoses
- Ceramidase
- Farber disease
- Krabbe disease
- Infantile onset
- Late onset
- Galactosialidosis
- Gangliosides: gangliosidoses
- Alpha-galactosidase
- Fabry disease (alpha-galactosidase A)
- Schindler disease (alpha-galactosidase B)
- Beta-galactosidase / GM1 gangliosidosis
- Infantile
- Juvenile
- Adult / chronic
- GM2 gangliosidosis
- AB variant
- Activator deficiency
- Sandhoff disease
- Infantile
- Juvenile
- Adult onset
- Tay–Sachs
- Juvenile hexosaminidase A deficiency
- Chronic hexosaminidase A deficiency
- Alpha-galactosidase
- Glucocerebroside
- Gaucher disease
- Type I
- Type II
- Type III
- Gaucher disease
- Sphingomyelinase
- Lysosomal acid lipase deficiency
- Early onset
- Late onset
- Niemann–Pick disease
- Type A
- Type B
- Lysosomal acid lipase deficiency
- Sulfatidosis
- Metachromatic leukodystrophy
- Saposin B deficiency
- Multiple sulfatase deficiency
- Metachromatic leukodystrophy
- Type I
- MPS I Hurler syndrome
- MPS I S Scheie syndrome
- MPS I H-S Hurler–Scheie syndrome
- Type II (Hunter syndrome)
- Type III (Sanfilippo syndrome)
- MPS III A (Type A)
- MPS III B (Type B)
- MPS III C (Type C)
- MPS III D (Type D)
- Type IV (Morquio)
- MPS IVA (Type A)
- MPS IVB (Type B)
- Type VI (Maroteaux–Lamy syndrome)
- Type VII (Sly syndrome)
- Type IX (hyaluronidase deficiency)
Mucolipidosis
- Type I (sialidosis)
- Type II (I-cell disease)
- Type III (pseudo-Hurler polydystrophy / phosphotransferase deficiency)
- Type IV (mucolipidin 1 deficiency)
- Niemann–Pick disease
- type C
- Type D
- Neuronal ceroid lipofuscinoses
- Type 1 Santavuori–Haltia disease / infantile NCL (CLN1 PPT1)
- Type 2 Jansky–Bielschowsky disease / late infantile NCL (CLN2/LINCL TPP1)
- Type 3 Batten–Spielmeyer–Vogt disease / juvenile NCL (CLN3)
- Type 4 Kufs disease / adult NCL (CLN4)
- Type 5 Finnish Variant / late infantile (CLN5)
- Type 6 Late infantile variant (CLN6)
- Type 7 CLN7
- Type 8 Northern epilepsy (CLN8)
- Type 8 Turkish late infantile (CLN8)
- Type 9 German/Serbian late infantile (unknown)
- Type 10 Congenital cathepsin D deficiency (CTSD)
- Wolman disease
Oligosaccharide
Lysosomal transport diseases
- Cystinosis
- Pycnodysostosis
- Salla disease / sialic acid storage disease
- Infantile free sialic acid storage disease
Glycogen storage diseases
- Type II Pompe disease
- Type IIb Danon disease[9]
Other
Lysosomal disease
Signs and symptoms
The symptoms of lysosomal storage diseases vary depending on the particular disorder and other variables such as the age of onset, and can be mild to severe. They can include developmental delay, movement disorders, seizures, dementia, deafness, and/or blindness. Some people with lysosomal storage diseases have enlarged livers or spleens, pulmonary and cardiac problems, and bones that grow abnormally.
Diagnosis
The majority of patients are initially screened by enzyme assay, which is the most efficient method to arrive at a definitive diagnosis. In some families where the disease-causing mutations are known, and in certain genetic isolates, mutation analysis may be performed. In addition, after a diagnosis is made by biochemical means, mutation analysis may be performed for certain disorders.
Treatment
No cures for lysosomal storage diseases are known, and treatment is mostly symptomatic, although bone marrow transplantation and enzyme replacement therapy (ERT) have been tried with some success.[11][12] ERT can minimize symptoms and prevent permanent damage to the body.[13] In addition, umbilical cord blood transplantation is being performed at specialized centers for a number of these diseases. In addition, substrate reduction therapy, a method used to decrease the production of storage material, is currently being evaluated for some of these diseases. Furthermore, chaperone therapy, a technique used to stabilize the defective enzymes produced by patients, is being examined for certain of these disorders. The experimental technique of gene therapy may offer cures in the future.[14][15]
Ambroxol has recently been shown to increase activity of the lysosomal enzyme glucocerebrosidase, so it may be a useful therapeutic agent for both Gaucher disease and Parkinson's disease.[16][17] Ambroxol triggers the secretion of lysosomes from cells by inducing a pH-dependent calcium release from acidic calcium stores.[18] Hence, relieving the cell from accumulating degradation products is a proposed mechanism by which this drug may help.
History
Tay–Sachs disease was the first of these disorders to be described, in 1881, followed by Gaucher disease in 1882. In the late 1950s and early 1960s, de Duve and colleagues, using cell fractionation techniques, cytological studies, and biochemical analyses, identified and characterized the lysosome as a cellular organelle responsible for intracellular digestion and recycling of macromolecules. This was the scientific breakthrough that would lead to the understanding of the physiological basis of the lysosomal storage diseases. Pompe disease was the first disease to be identified as an lysosomal storage disease in 1963, with L. Hers reporting the cause as a deficiency of α-glucosidase. Hers also suggested that other diseases, such as the mucopolysaccharidosis, might be due to enzyme deficiencies.
See also
References
- Platt, Frances M.; d’Azzo, Alessandra; Davidson, Beverly L.; Neufeld, Elizabeth F.; Tifft, Cynthia J. (2018-10-01). "Lysosomal storage diseases". Nature Reviews Disease Primers. 4 (1): 27. doi:10.1038/s41572-018-0025-4. ISSN 2056-676X. PMID 30275469. S2CID 52896843.
- Winchester B, Vellodi A, Young E (2000). "The molecular basis of lysosomal storage diseases and their treatment". Biochem. Soc. Trans. 28 (2): 150–4. doi:10.1042/bst0280150. PMID 10816117.
- Reece, Jane; Campbell, Neil (2002). Biology. San Francisco: Benjamin Cummings. pp. 121–122. ISBN 0-8053-6624-5.
- Meikle, P. J.; Hopwood, J. J.; Clague, A. E.; Carey, W. F. (20 January 1999). "Prevalence of lysosomal storage disorders". JAMA. 281 (3): 249–254. doi:10.1001/jama.281.3.249. ISSN 0098-7484. PMID 9918480.
- M, Fuller; PJ, Meikle; JJ, Hopwood (1 January 2006). "Epidemiology of lysosomal storage diseases: an overview". PMID 21290699.
{{cite journal}}: Cite journal requires|journal=(help) - eMedicine Specialties > Neurology > Pediatric Neurology > Lysosomal Storage Disease Author: Noah S Scheinfeld, MD, JD, FAAD. Coauthor(s): Rowena Emilia Tabamo, MD; Brian Klein, MD. Updated: Sep 25, 2008
- Medical Physiology (2nd Edition) – W. Boron & E. Boulpaep, Saunders Press
- Table 7-6 in:Mitchell, Richard Sheppard; Kumar, Vinay; Abbas, Abul K.; Fausto, Nelson (2007). Robbins Basic Pathology. Philadelphia: Saunders. ISBN 978-1-4160-2973-1. 8th edition.
- "Danon disease".
- Clarke JT, Iwanochko RM (2005). "Enzyme replacement therapy of Fabry disease". Mol. Neurobiol. 32 (1): 043–050. doi:10.1385/MN:32:1:043. PMID 16077182. S2CID 24240533.
- Bruni S, Loschi L, Incerti C, Gabrielli O, Coppa GV (2007). "Update on treatment of lysosomal storage diseases". Acta Myol. 26 (1): 87–92. PMC 2949325. PMID 17915580.
- "Enzyme Replacement Therapy for Gaucher Disease". National Gaucher Foundation. Retrieved 2017-06-08.
- Nagree, Murtaza S.; Scalia, Simone; McKillop, William M.; Medin, Jeffrey A. (2019-07-03). "An update on gene therapy for lysosomal storage disorders". Expert Opinion on Biological Therapy. 19 (7): 655–670. doi:10.1080/14712598.2019.1607837. ISSN 1471-2598. PMID 31056978. S2CID 145822883.
- Ponder KP, Haskins ME (2007). "Gene therapy for mucopolysaccharidosis". Expert Opin Biol Ther. 7 (9): 1333–1345. doi:10.1517/14712598.7.9.1333. PMC 3340574. PMID 17727324.
- McNeill, Alisdair; Magalhaes, Joana; Shen, Chengguo; Chau, Kai-Yin; Hughes, Derralyn; Mehta, Atul; Foltynie, Tom; Cooper, J. Mark; Abramov, Andrey Y. (2014-05-01). "Ambroxol improves lysosomal biochemistry in glucocerebrosidase mutation-linked Parkinson disease cells". Brain. 137 (5): 1481–1495. doi:10.1093/brain/awu020. ISSN 0006-8950. PMC 3999713. PMID 24574503.
- Albin, Roger L.; Dauer, William T. (2014-05-01). "Magic shotgun for Parkinson's disease?". Brain. 137 (5): 1274–1275. doi:10.1093/brain/awu076. ISSN 0006-8950. PMID 24771397.
- Fois, Giorgio; Hobi, Nina; Felder, Edward; Ziegler, Andreas; Miklavc, Pika; Walther, Paul; Radermacher, Peter; Haller, Thomas; Dietl, Paul (2015). "A new role for an old drug: Ambroxol triggers lysosomal exocytosis via pH-dependent Ca2+ release from acidic Ca2+ stores". Cell Calcium. 58 (6): 628–637. doi:10.1016/j.ceca.2015.10.002. PMID 26560688.