Sandhoff disease
Sandhoff disease is a lysosomal genetic, lipid storage disorder caused by the inherited deficiency to create functional beta-hexosaminidases A and B.[1][2] These catabolic enzymes are needed to degrade the neuronal membrane components, ganglioside GM2, its derivative GA2, the glycolipid globoside in visceral tissues,[1] and some oligosaccharides. Accumulation of these metabolites leads to a progressive destruction of the central nervous system and eventually to death.[1][3] The rare autosomal recessive[4][5] neurodegenerative disorder is clinically almost indistinguishable from Tay–Sachs disease, another genetic disorder that disrupts beta-hexosaminidases A and S. There are three subsets of Sandhoff disease based on when first symptoms appear: classic infantile, juvenile and adult late onset.
| Sandhoff disease | |
|---|---|
| Other names | Sandhoff–Jatzkewitz disease, variant 0 of GM2-gangliosidosis or hexosaminidase A and B deficiency |
 | |
| Sandhoff disease is inherited via an autosomal recessive manner. | |
| Specialty | Endocrinology |
Symptoms and signs
Sandhoff disease symptoms are clinically indeterminable from Tay–Sachs disease. The classic infantile form of the disease has the most severe symptoms and is incredibly hard to diagnose at this early age.[6] The first signs of symptoms begin before 6 months of age and the parents’ notice when the child begins regressing in their development. If the children had the ability to sit up by themselves or crawl they will lose this ability. This is caused by a slow deterioration of the muscles in the child’s body from the buildup of GM2 gangliosides. Since the body is unable to create the enzymes it needs within the central nervous system, it is unable to attach to these gangliosides to break them apart and make them non-toxic. With this buildup there are several symptoms that begin to appear such as muscle/motor weakness, sharp reaction to loud noises, blindness, deafness, inability to react to stimulants, respiratory problems and infections, mental retardation, seizures, cherry red spots in the retina, enlarged liver and spleen (hepatosplenomegaly), pneumonia, or bronchopneumonia.[7]
The other two forms of Sandhoff disease have similar symptoms but to a lesser extent. Adult and juvenile forms of Sandhoff disease are more rare than the infantile form.[8] In these cases victims suffer cognitive impairment (retardation) and a loss of muscle coordination that impairs and eventually destroys their ability to walk; the characteristic red spots in the retina also develop. The adult form of the disease, however, is sometimes milder, and may only lead to muscle weakness that impairs walking or the ability to get out of bed.[9]
Causes
Two parents carrying a mutated gene and passing it on to their offspring cause the disease. Even with both parents carrying the disease in their genome, there is only a 25% chance that they will have a child containing the genetic coding for the disease (see figure right).[10]
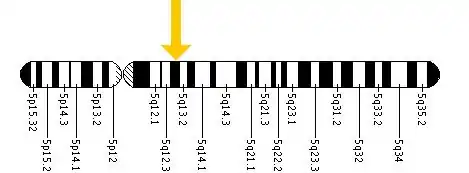
Each form of the disease is caused by the differences in the various mutations of the genome, in particular the codons on the 14 exons in the HEX B gene located within chromosome 5 (see figure bottom), leading to the differences in severities of the symptoms.[6] The difference in the codons has the consequence of inhibiting two enzymes located in the lysosomes of the neurons of the central nervous system. Lysosomes contain various enzymes to break down byproducts and toxins to ensure they do not accumulate enough to interfere with the function of the central nervous system.[7]
Using restriction enzymes, it was discovered that a mutation on chromosome 5 particularly within the C1214T allele caused the adult onset form of Sandhoff Disease. For the patient showing symptoms of the infantile or juvenile form they have a mutation on exon I207V from their father, and a 16 base pair deletion from their mother which can be located on as many as five exons, exons 1–5.[11]
Mutations and polymorphism
Articles regarding Sandhoff disease frequencies among distinct groups of people contain discrepancies from one another. More than 25 mutations have been reported other than novel mutations.[5]
One article says that Sandhoff disease is found commonly in individuals with a non-Jewish descent.[12]
Others say that it is more commonly in:
- the Creole population of northern Argentina[13]
- the indigenous Métis in Saskatchewan[10]
- Christian Maronite communities from Cyprus[14]
Discovery of several mutations in Ashkenazi Jews may reflect ascertainment bias rather than a high population frequency, because Ashkenazi Jews were the targeted population in a mass screening program for Tay-Sachs disease. Several rare SD mutations were discovered as researchers resolved cases of enzyme deficiency among suspected TSD carriers, but no cases of the disease itself have been reported.[5][15]
However, since it is an autosomal recessive disease, it is likely found in any ethnic group passing from generation to generation through carriers without being expressed in their offspring. Even though the family may not have a history of Sandhoff disease, it is possible for two individuals to have a child with the disease. Since Sandhoff disease was only discovered in 1968, there are years the disease has gone undetected because of misdiagnoses.
Pathophysiology
Biallelic pathogenic variants in the HEXB gene cause Sandhoff disease. The gene provides instructions for making a protein crucial to the enzymes beta-hexosaminidase A and beta-hexosaminidase B,[16] which function in nerve cells to break down fatty substances, complex sugars, and molecules that are linked to sugars. In particular, beta-hexosaminidase A breaks down a fatty compound called GM2 ganglioside. Mutations in the HEXB gene disrupt the activity of these enzymes, preventing the breakdown of GM2 ganglioside and other molecules.
As a result, progressive damage caused by the resulting buildup of GM2 ganglioside leads to the destruction of nerve cells, causing the signs and symptoms associated with Sandhoff disease.
Diagnosis
Sandhoff disease can be detected through the following procedures (before it is apparent through physical examination): a biopsy removing a sample of tissue from the liver, genetic testing, molecular analysis of cells and tissues (to determine the presence of a genetic metabolic disorder), enzyme assay, and occasionally a urinalysis to determine if the above-noted compounds are abnormally stored within the body. For a child to suffer from this disease, both parents must be carriers, and both must transmit the mutation to the child. Thus, even in the case where both parents have the mutation, there is only a 25 percent chance their child will inherit the condition. Frequently, parents are given the opportunity to have a DNA screening if they are at high risk, to determine their carrier status before they have children. However, it is also highly recommended to undergo testing even for those parents who do not have a family history of Sandhoff disease. Over 95% of the families that have children with Sandhoff disease had no known prior family history of the condition, as the mutation in the HEXB gene does not cause clinical symptoms when only one copy is present, and often passed undetected from one generation to the next[6] Naturally, if an individual carries the mutation, he or she has a risk of transmitting it to the unborn child. Genetic counseling is recommended for those who have the mutation.
It is possible for parents who are about to have a child or had a child with Sandhoff Disease can have a PGD or PEGD. PEGD is pre-embryonic genetic diagnosis for the parents that would not benefit from a pre-implantation genetic diagnosis because of their religion or negative attitude for the discarding of embryos. PEGD sequences the genome of the embryo to be produced by two parents if they were to conceive a child. If the family has a history of Sandhoff disease it is recommended they have their genome sequenced to ensure they are not carriers or to sequence the genome of their child.[17]
Types
There are three types of Sandhoff disease: classic infantile, juvenile, and adult late onset.[16] Each form is classified by the severity of the symptoms as well as the age at which the patient shows these symptoms.[18]
- Classic infantile form of the disease is classified by the development of symptoms anywhere from 2 months to 9 months of age. It is the most common and most severe of all of the forms and will lead to death before the patient reaches the age of three.[19] Infants with this disorder typically appear normal until the age of 3 to 6 months, when development slows and muscles used for movement weaken. Affected infants lose motor skills such as turning over, sitting, and crawling. As the disease progresses, infants develop seizures, vision and hearing loss, dementia, and paralysis. An eye abnormality called a cherry-red spot, which can be identified with an eye examination, is characteristic of this disorder. Some infants with Sandhoff disease may have enlarged organs (organomegaly) or bone abnormalities. Children with the severe form of this disorder usually live only into early childhood.
- The juvenile form of the disease shows symptoms starting at age 3 ranging to age 10 and, although the child usually dies by the time they are 15, it is possible for them to live longer if they are under constant care.[20] Symptoms include autism, ataxia, motor skills regression, spacticity, and learning disorders.[21]
- Adult onset form of the disease is classified by its occurrence in older individuals and has an effect on the motor function of these individuals. It is not yet known if Sandhoff disease will cause these individuals to have a decrease in their life span.[6]
Juvenile and adult onset forms of Sandhoff disease are very rare. Signs and symptoms can begin in childhood, adolescence, or adulthood and are usually milder than those seen with the infantile form of Sandhoff disease. As in the infantile form, mental abilities and coordination are affected. Characteristic features include muscle weakness, loss of muscle coordination (ataxia) and other problems with movement, speech problems, and mental illness. These signs and symptoms vary widely among people with late-onset forms of Sandhoff disease.
Treatment
Currently Sandhoff disease does not have any standard treatment and does not have a cure. However, a person suffering from the disease needs proper nutrition, hydration, and maintenance of clear airways. To reduce some symptoms that may occur with Sandhoff disease, the patient may take anticonvulsants to manage seizures or medications to treat respiratory infections, and consume a precise diet consisting of puree foods due to difficulties swallowing. Infants with the disease usually die by the age of 3 due to respiratory infections. The patient must be under constant surveillance because they can suffer from aspiration or lack the ability to change from the passageway to their lungs versus their stomach and their spit travels to the lungs causing bronchopneumonia. The patient also lacks the ability to cough and therefore must undergo a treatment to shake up their body to remove the mucus from the lining of their lungs. Medication is also given to patients to lessen their symptoms including seizures.
Currently the government is testing several treatments including N-butyl-deoxynojirimycin in mice, as well as stem cell treatment in humans and other medical treatments recruiting test patients.[11] A Sandhoff disease study showing proof of principle for gene therapy in a human model system using CRISPR and virus gene correction gives the chance for clinical trials to cure the disease. The ultra-rare occurrence is a main hurdle to overcome for clinical trials.[22][23]
History

Sandhoff disease is one of several forms of what was formerly known as amaurotic idiocy. This inherited disease is characterized by the accumulation of lipid-containing cells in the viscera and in the nervous system, mental retardation, and impaired vision or blindness. The chemical and enzymatic analysis of various patients with amaurotic idiocy by Konrad Sandhoff (1939- ), a German biochemist, led to the identification of several biochemically distinct diseases: The first biochemical description of GM1-gangliosidosis in 1963,[24] Sandhoff disease in 1968,[1] Tay-Sachs-Disease,[2][25] the AB-variant of GM2-Gangliosidosis[2][26] and the B1-variant of GM2-gangliosidosis.[27]
The molecular defect in Sandhoff disease was discovered when Konrad Sandhoff studied the biochemistry of sphingolipids and gangliosides in the laboratory of Prof. Horst Jatzkewitz (1912–2002), a German biochemist (Max- Planck-Institute for Psychiatry, Munich). In October 1966, he obtained deep-frozen autopsy material from an infantile case with amaurotic idiocy. The glycolipid analysis soon demonstrated differences from all the cases studied before. Besides the neuronal storage of GM2, the storage of GA2 was much more pronounced, and different from all cases of Tay-Sachs disease studied so far, globoside accumulated in the visceral organs and, most importantly, hexosaminidase activity was almost completely absent. The disease causing catabolic enzyme deficiency of hexosaminidases was demonstrated with four different substrates (p–nitrophenyl-β-D-N-acetylglucosaminide, p-nitrophenyl-β-D-N-acetylgalactosaminide, glycolipid [3H]GA2 and [3H]globoside) in four different organs and published in 1968.[1]
See also
- GM2-gangliosidosis, AB variant
- globoside
References
- Sandhoff K, Andreae U, Jatzkewitz H (March 1968). "Deficient hexosaminidase activity in an exceptional case of Tay-Sachs disease with additional storage of kidney globoside in visceral organs". Life Sci. 7 (6): 283–8. doi:10.1016/0024-3205(68)90024-6. PMID 5651108.
- Sandhoff K (August 1969). "Variation of beta-N-acetylhexosaminidase-pattern in Tay-Sachs disease". FEBS Lett. 4 (4): 351–354. doi:10.1016/0014-5793(69)80274-7. PMID 11947222. S2CID 84542601.
- Pilz H, Müller D, Sandhoff K, ter Meulen V (September 1968). "Tay-Sachssche Krankheit mit Hexosaminidase-Defekt (Klinische, morphologische und biochemische Befunde bei einem Fall mit viszeraler Speicherung von Nierenglobosid)". Dtsch Med Wochenschr. 93 (39): 1833–9. doi:10.1055/s-0028-1110836. PMID 5679107.
- Harzer K, Sandhoff K, Schall H, Kollmann F (November 1971). "Enzymatische Untersuchungen im Blut von Überträgern einer Variante der Tay-Sachsschen Erkrankung (Variante 0)". Klin Wochenschr. 49 (21): 1189–91. doi:10.1007/bf01732464. PMID 5124584. S2CID 1735733.
- Online Mendelian Inheritance in Man (OMIM): Sandhoff Disease - 268800
- Gomez-Lira M, Sangalli A, Mottes M, Perusi C, Pignatti PF, Rizzuto N, Salviati A (1995). "A common β hexosaminidase gene mutation in adult Sandhoff disease patients". Human Genetics. 96 (4): 417–422. doi:10.1007/bf00191799. PMID 7557963. S2CID 39688704.
- "Introduction to Sandhoff Disease". The Medical Biochemistry Page. Retrieved 2009-05-03.
- "Sandhoff Disease". Genetics Home Reference. Retrieved 2009-05-03.
- "Symptoms of Sandhoff Disease". Medical Books Excerpts. Lippincott Williams & Wilkin. 2008.
- Lowden JA, et al. (1978). "Carrier detection in Sandhoff disease". American Journal of Human Genetics. 30 (1): 338–345. PMC 1685463. PMID 414620.
- "Lysosomal Diseases Testing Laboratory". Department of Neurology Jefferson Hospital. Archived from the original on April 10, 2009. Retrieved 2009-05-03.
- "Carrier Testing". National Tay-Sachs & Allied Disease Association, Inc. Retrieved 2009-05-03.
- Kleiman FE, et al. (1994). "Sandhoff disease in Argentina: high frequency of a splice site mutation in the HEXB gene and correlation between enzyme and DNA-based tests for heterozygote detection". Human Genetics. 94 (3): 279–82. doi:10.1007/bf00208283. PMID 8076944. S2CID 9666991.
- Drousiotou A, et al. (2000). "Sandhoff disease in Cyprus: population screening by biochemical and DNA analysis indicates a high frequency of carriers in the Maronite community". Human Genetics. 107 (1): 12–17. doi:10.1007/s004390050003. PMID 10982028.
- Cantor RM, Kaback MM (1985). "Sandhoff disease (SHD) heterozygote frequencies (HF) in North American (NA) Jewish (J) and non-Jewish (NJ) populations: implications for carrier (C) screening". American Journal of Human Genetics. 37: A48.
- Chamoles NA, Blanco M, Gaggioli D, Casentini C (April 2002). "Tay-Sachs and Sandhoff diseases: enzymatic diagnosis in dried blood spots on filter paper: retrospective diagnoses in newborn-screening cards". Clinica Chimica Acta. 318 (1–2): 133–7. doi:10.1016/S0009-8981(02)00002-5. PMID 11880123.
- Kuliev A, Rechitsky S, Laziuk K, Verlinsky O, Tur-Kaspa I, Verlinsky Y (2006). "Pre-Embryonic diagnosis for Sandhoff Disease". Reproductive BioMedicine Online. 12 (3): 328–333. doi:10.1016/S1472-6483(10)61005-X. PMID 16569321.
- Zhang, Zhi-Xin; Nobuaki Wakamatsu; Emilie H. Mulesi; George H. Thomasi; Roy A. Gravel (1994). "Impact of premature stop codons on mRNA levels in infantile Sandhoff disease". Human Molecular Genetics. 3 (1): 139–145. doi:10.1093/hmg/3.1.139. PMID 8162015.
- "From a parents perspective: Parents view of Sandhoff". sandhoffdisease.webs.com. Archived from the original on 2009-01-29. Retrieved 2009-05-03.
- Hendriksz CJ, Corry PC, Wraith JE, Besley GT, Cooper A, Ferrie CD (2004). "Juvenile Sandhoff disease-Nine New Cases and a review of the literature". Journal of Inherited Metabolic Disease. 27 (2): 241–9. doi:10.1023/B:BOLI.0000028777.38551.5a. PMID 15159655. S2CID 41447979.
- Karbani, Gulshan A (15 May 2012). "Genetic Counselling: Consanguinity and Cultural Expectations". Encyclopedia Of Life Sciences. doi:10.1002/9780470015902.a0006179.pub2. ISBN 978-0470016176.
- Allende, Maria L.; Cook, Emily K.; Larman, Bridget C.; Nugent, Adrienne; Brady, Jacqueline M.; Golebiowski, Diane; Sena-Esteves, Miguel; Tifft, Cynthia J.; Proia, Richard L. (2018-01-22). "Cerebral organoids derived from Sandhoff disease induced pluripotent stem cells exhibit impaired neurodifferentiation". Journal of Lipid Research. 59 (3): 550–563. doi:10.1194/jlr.M081323. ISSN 0022-2275. PMC 5832932. PMID 29358305.
- "Sandhoff disease study shows proof of principle for gene therapy - Scienmag: Latest Science and Health News". Scienmag: Latest Science and Health News. 2018-02-22. Retrieved 2018-02-23.
- Jatzkewitz H, Sandhoff K (June 1963). "On a biochemically special form of infantile amaturotic idiocy". Biochim Biophys Acta. 70: 354–6. doi:10.1016/0006-3002(63)90764-9. PMID 13957544.
- Okada S, O'Brien JS (August 1969). "Tay-Sachs disease: generalized absence of a beta-D-N-acetylhexosaminidase component". Science. 165 (894): 698–700. Bibcode:1969Sci...165..698O. doi:10.1126/science.165.3894.698. PMID 5793973. S2CID 8473726.
- Conzelmann E, Sandhoff K (August 1978). "AB variant of infantile GM2 gangliosidosis: deficiency of a factor necessary for stimulation of hexosaminidase A-catalyzed degradation of ganglioside GM2 and glycolipid GA2". Proc Natl Acad Sci U S A. 75 (8): 3979–83. Bibcode:1978PNAS...75.3979C. doi:10.1073/pnas.75.8.3979. PMC 392913. PMID 99746.
- Kytzia HJ, Hinrichs U, Maire I, Suzuki K, Sandhoff K (1983). "Variant of GM2-gangliosidosis with hexosaminidase A having a severely changed substrate specificity". EMBO J. 2 (7): 1201–5. doi:10.1002/j.1460-2075.1983.tb01567.x. PMC 555256. PMID 6226523.
This article incorporates some public domain text from The U.S. National Library of Medicine