Lysosomal acid lipase deficiency
Lysosomal acid lipase deficiency (LAL deficiency or LAL-D) is an autosomal recessive inborn error of metabolism that results in the body not producing enough active lysosomal acid lipase (LAL) enzyme. This enzyme plays an important role in breaking down fatty material (cholesteryl esters and triglycerides) in the body.[1] Infants, children and adults that have LAL deficiency experience a range of serious health problems. The lack of the LAL enzyme can lead to a build-up of fatty material in a number of body organs including the liver, spleen, gut, in the wall of blood vessels and other important organs.
| Lysosomal acid lipase deficiency | |
|---|---|
| Other names | Wolman disease |
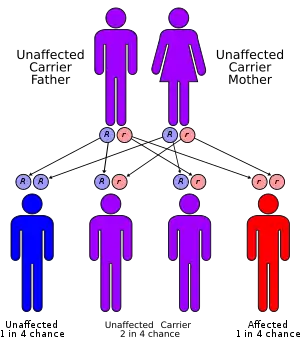 | |
| LAL-D has an autosomal recessive pattern of inheritance. | |
| Specialty | Medical Genetics, Hepatology |
| Usual onset | congenital |
Very low levels of the LAL enzyme lead to LAL deficiency. LAL deficiency typically affects infants in the first year of life. The accumulation of fat in the walls of the gut in early onset disease leads to serious digestive problems including malabsorption, a condition in which the gut fails to absorb nutrients and calories from food. Because of these digestive complications, affected infants usually fail to grow and gain weight at the expected rate for their age (failure to thrive). As the disease progresses, it can cause life-threatening liver dysfunction or liver failure.[2]
Until 2015, there was no treatment, and very few infants with LAL-D survived beyond the first year of life. In 2015, an enzyme replacement therapy, sebelipase alfa, was approved in the US and EU. The therapy was additionally approved in Japan in 2016.
Symptoms and signs
Infants may present with feeding difficulties with frequent vomiting, diarrhea, swelling of the abdomen, and failure to gain weight or sometimes weight loss.[3]
As the disease progresses in infants, increasing fat accumulation in the liver leads to other complications including yellowing of the skin and whites of the eyes (jaundice), and a persistent low-grade fever. An ultrasound examination shows accumulation of chalky material (calcification) in the adrenal gland in about half of infants with LAL-D.[3][4] Complications of LAL-D progress over time, eventually leading to life-threatening problems such as extremely low levels of circulating red blood cells (severe anemia), liver dysfunction or failure, and physical wasting (cachexia).[3]
People who are older children or adults generally present with a wide range of signs and symptoms that overlap with other disorders.[5] They may have diarrhoea, stomach pain, vomiting, or poor growth, a sign of malabsorption. They may have signs of bile duct problems, like itchiness, jaundice, pale stool, or dark urine. Their feces may be excessively greasy. They often have an enlarged liver, liver disease, and may have yellowish deposits of fat underneath the skin, usually around their eyelids.[3][5] The disease is often undiagnosed in adults.[6] The person may have a history of premature cardiac disease or premature stroke.[3]
Cause
Lysosomal acid lipase deficiency is a genetic disease that is autosomal recessive. It is an inborn error of metabolism that causes a lysosomal storage disease.[3] The condition is caused by a mutation of the LIPA gene, which is responsible for the gene coding of the lysosomal lipase protein (also called lysosomal acid lipase or LAL), which results in a loss of the protein's normal function.[2] When LAL functions normally, it breaks down cholesteryl esters and triglycerides in low density lipoprotein particles into free cholesterol and free fatty acids that the body can reuse; when LAL doesn't function, cholesteryl esters and triglycerides build up in the liver, spleen and other organs.[3][5] The accumulation of fat in the walls of the gut and other organs in leads to serious digestive problems including malabsorption, a condition in which the gut fails to absorb nutrients and calories from food, persistent and often forceful vomiting, frequent diarrhea, foul-smelling and fatty stools (steatorrhea), and failure to grow.[3]
Lysosomal acid lipase deficiencies occur when a person has defects (mutations) in both copies of the LIPA gene. Each parent of a person with LAL deficiency carries one copy of the defective LIPA gene. With every pregnancy, parents with a son or daughter affected by LAL deficiency have a 1 in 4 (25%) chance of having another affected child. A person born with defects in both LIPA genes is not able to produce adequate amounts of the LAL enzyme.[5]
Diagnosis
Blood tests may show anaemia and their lipid profiles are generally similar to people with more common familial hypercholesterolemia, including elevated total cholesterol, elevated low-density lipoprotein cholesterol, decreased high-density lipoprotein cholesterol and elevated serum transaminases.[3]
Liver biopsy findings will generally show a bright yellow-orange color, enlarged, lipid-laden hepatocytes and Kupffer cells, microvesicular and macrovesicular steatosis, fibrosis, and cirrhosis.[3] The only definitive tests are genetic, which may be conducted in any number of ways.[5]
Screening
Because LAL deficiency is inherited, each sibling of an affected individual has a 25% chance of having pathological mutations in LAL genes from both their mother and their father, a 50% chance of having a pathological mutation in only one gene, and a 25% chance of having no pathological mutations. Genetic testing for family members and genetic prenatal diagnosis of pregnancies for women who are at increased risk are possible if family members carrying pathological mutations have been identified.[5]
Management
LAL deficiency can be treated with sebelipase alfa is a recombinant form of LAL that was approved in 2015 in the US and EU.[7][8] The disease of LAL affects < 0.2 in 10,000 people in the EU.[8] According to an estimate by a Barclays analyst, the drug will be priced at about US$375,000 per year.[8]
It is administered once a week via intraveneous infusion in people with rapidly progressing disease in the first six months of life. In people with less aggressive disease, it is given every other week.[9]
Before the drug was approved, treatment of infants was mainly focused on reducing specific complications and was provided in specialized centers. Specific interventions for infants included changing from breast or normal bottle formula to a specialized low fat formula, intravenous feeding, antibiotics for infections, and steroid replacement therapy because of concerns about adrenal function.[3]
Statins were used in people with LAL-D prior to the approval of sebelipase alfa; they helped control cholesterol but did not appear to slow liver damage; liver transplantation was necessary in most patients.[3]
Prognosis
Infants with LAL deficiencies typically show signs of disease in the first weeks of life and if untreated, die within 6–12 months due to multi-organ failure.[3] Older children or adults with LAL-D may remain undiagnosed or be misdiagnosed until they die early from a heart attack or stroke or die suddenly of liver failure.[3] The first enzyme replacement therapy was approved in 2015. In those clinical trials nine infants were followed for one year; 6 of them lived beyond one year.[9] Older children and adults were followed for 36 weeks.[9]
Epidemiology
Depending on ethnicity and geography, prevalence has been estimated to be between 1 in 40,000 and 1 in 300,000; based on these estimates the disease may be underdiagnosed. Jewish infants of Iraqi or Iranian origin appear to be most at risk based on a study of a community in Los Angeles in which there was a prevalence of 1 in 4200.[3][5]
History
In 1956, Moshe Wolman, along with two other doctors, published the first case study of a LAL deficiency in a child born to closely related Persian Jews; 12 years later a case study on an older boy was published, which turned out to be the first case study of LAL-D.[3][10][11][12]
LAL-D was historically referred to as two separate disorders:
- Wolman disease, presenting in infant patients
- Cholesteryl Ester Storage Disease, presenting in pediatric and adult patients
Around 2010 both presentations have come to be known as LAL-D, as both are due to a deficiency of the LAL enzyme.[2]
In 2015 an enzyme replacement therapy, sebelipase alfa, was approved in the US and EU for the treatment of human LAL enzyme deficiency.[13] Before the approval of that drug, as of 2009 the two oldest survivors of LAL-D in the world were then aged 4 and 11; both of them had been treated with hematopoietic stem cell treatment.[14]
Research directions
Some children with LAL-D have had an experimental therapy called hematopoietic stem cell transplantation (HSCT), also known as bone marrow transplant, to try to prevent the disease from getting worse. Data are sparse but there is a known high risk of serious complications including death, graft-versus-host disease.[3]
References
- "Wolman disease". Genetics Home Reference. 2016-03-21. Retrieved 2016-03-25.
- Reiner, Željko; Guardamagna, Ornella; Nair, Devaki; Soran, Handrean; Hovingh, Kees; Bertolini, Stefano; Jones, Simon; Ćorić, Marijana; Calandra, Sebastiano; Hamilton, John; Eagleton, Terence; Ros, Emilio (July 2014). "Lysosomal acid lipase deficiency – An under-recognized cause of dyslipidaemia and liver dysfunction". Atherosclerosis. 235 (1): 21–30. doi:10.1016/j.atherosclerosis.2014.04.003. PMID 24792990.
- Reiner Ž; et al. (Jul 2014). "Lysosomal acid lipase deficiency--an under-recognized cause of dyslipidaemia and liver dysfunction". Atherosclerosis. 235 (1): 21–30. doi:10.1016/j.atherosclerosis.2014.04.003. PMID 2479299.
- Learning Radiology.com Adrenal calcification
- Hoffman EP, Barr ML, Giovanni MA, et al. Lysosomal Acid Lipase Deficiency. 2015 Jul 30. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2016.
- Bernstein, Donna L.; Hülkova, Helena; Bialer, Martin G.; Desnick, Robert J. (Jun 2013). "Cholesteryl ester storage disease: Review of the findings in 135 reported patients with an underdiagnosed disease". Journal of Hepatology. 58 (6): 1230–1243. doi:10.1016/j.jhep.2013.02.014. PMID 23485521.
- Burton, B. K.; et al. (September 10, 2015). "A Phase 3 Trial of Sebelipase Alfa in Lysosomal Acid Lipase Deficiency". New England Journal of Medicine. 373 (11): 1010–1020. doi:10.1056/NEJMoa1501365. PMID 26352813.
- "New Drugs Online Report for sebelipase alfa". UK Medicines Information. Archived from the original on March 4, 2016. Retrieved December 10, 2015.
- Sebelipase alfa Label Last updated Dec 2015. See FDA index page for labels here
- synd/3122 at Who Named It?
- Abramov A, Schorr S, Wolman M (Mar 1956). "Generalized xanthomatosis with calcified adrenals". AMA J Dis Child. 91 (3): 282–6. doi:10.1001/archpedi.1956.02060020284010. PMID 13301142.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - Fredrickson DS (1963). "Newly recognized disorders of cholesterol metabolism". Ann Intern Med. 58 (4): 718. doi:10.7326/0003-4819-58-4-718_1.
- "FDA approves first drug to treat a rare enzyme disorder in pediatric and adult patients". FDA. December 8, 2015. Retrieved December 10, 2015.
- Tolar, J.; Petryk, A.; Khan, K.; Bjoraker, K. J.; Jessurun, J.; Dolan, M.; Kivisto, T.; Charnas, L.; Shapiro, E. G. (2009-01-01). "Long-term metabolic, endocrine, and neuropsychological outcome of hematopoietic cell transplantation for Wolman disease". Bone Marrow Transplantation. 43 (1): 21–27. doi:10.1038/bmt.2008.273. ISSN 1476-5365. PMID 18776925.
External links
- National Organization for Rare Disorders (NORD)
- Article - LYSOSOMAL ACID LIPASE/NIH.gov
- Article - LYSOSOMAL ACID LIPASE DEFICIENCY/NIH.gov
- Lipid Storage Diseases Fact Sheet at ninds.nih.gov