Immunoprecipitation
Immunoprecipitation (IP) is the technique of precipitating a protein antigen out of solution using an antibody that specifically binds to that particular protein. This process can be used to isolate and concentrate a particular protein from a sample containing many thousands of different proteins. Immunoprecipitation requires that the antibody be coupled to a solid substrate at some point in the procedure.
Types
Individual protein immunoprecipitation (IP)
Involves using an antibody that is specific for a known protein to isolate that particular protein out of a solution containing many different proteins. These solutions will often be in the form of a crude lysate of a plant or animal tissue. Other sample types could be body fluids or other samples of biological origin.
Protein complex immunoprecipitation (Co-IP)
Immunoprecipitation of intact protein complexes (i.e. antigen along with any proteins or ligands that are bound to it) is known as co-immunoprecipitation (Co-IP). Co-IP works by selecting an antibody that targets a known protein that is believed to be a member of a larger complex of proteins. By targeting this known member with an antibody it may become possible to pull the entire protein complex out of solution and thereby identify unknown members of the complex.
This works when the proteins involved in the complex bind to each other tightly, making it possible to pull multiple members of the complex out of solution by latching onto one member with an antibody. This concept of pulling protein complexes out of solution is sometimes referred to as a "pull-down". Co-IP is a powerful technique that is used regularly by molecular biologists to analyze protein–protein interactions.
- A particular antibody often selects for a subpopulation of its target protein that has the epitope exposed, thus failing to identify any proteins in complexes that hide the epitope. This can be seen in that it is rarely possible to precipitate even half of a given protein from a sample with a single antibody, even when a large excess of antibody is used.
- As successive rounds of targeting and immunoprecipitations take place, the number of identified proteins may continue to grow. The identified proteins may not ever exist in a single complex at a given time, but may instead represent a network of proteins interacting with one another at different times for different purposes.
- Repeating the experiment by targeting different members of the protein complex allows the researcher to double-check the result. Each round of pull-downs should result in the recovery of both the original known protein as well as other previously identified members of the complex (and even new additional members). By repeating the immunoprecipitation in this way, the researcher verifies that each identified member of the protein complex was a valid identification. If a particular protein can only be recovered by targeting one of the known members but not by targeting other of the known members then that protein's status as a member of the complex may be subject to question.
Chromatin immunoprecipitation (ChIP)
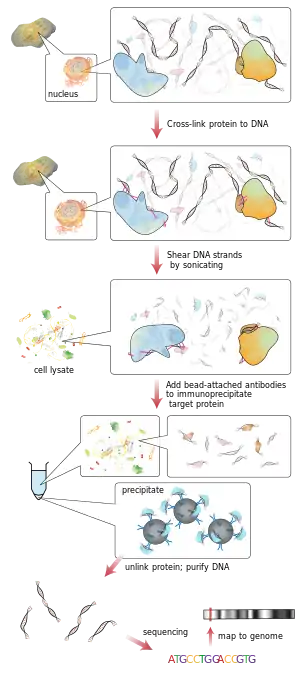
Chromatin immunoprecipitation (ChIP) is a method used to determine the location of DNA binding sites on the genome for a particular protein of interest. This technique gives a picture of the protein–DNA interactions that occur inside the nucleus of living cells or tissues. The in vivo nature of this method is in contrast to other approaches traditionally employed to answer the same questions.
The principle underpinning this assay is that DNA-binding proteins (including transcription factors and histones) in living cells can be cross-linked to the DNA that they are binding. By using an antibody that is specific to a putative DNA binding protein, one can immunoprecipitate the protein–DNA complex out of cellular lysates. The crosslinking is often accomplished by applying formaldehyde to the cells (or tissue), although it is sometimes advantageous to use a more defined and consistent crosslinker such as dimethyl 3,3′-dithiobispropionimidate-2 HCl (DTBP).[1] Following crosslinking, the cells are lysed and the DNA is broken into pieces 0.2–1.0 kb in length by sonication. At this point the immunoprecipitation is performed resulting in the purification of protein–DNA complexes. The purified protein–DNA complexes are then heated to reverse the formaldehyde cross-linking of the protein and DNA complexes, allowing the DNA to be separated from the proteins. The identity and quantity of the DNA fragments isolated can then be determined by polymerase chain reaction (PCR). The limitation of performing PCR on the isolated fragments is that one must have an idea which genomic region is being targeted in order to generate the correct PCR primers. Sometimes this limitation is circumvented simply by cloning the isolated genomic DNA into a plasmid vector and then using primers that are specific to the cloning region of that vector. Alternatively, when one wants to find where the protein binds on a genome-wide scale, ChIP-sequencing is used and has recently emerged as a standard technology that can localize protein binding sites in a high-throughput, cost-effective fashion, allowing also for the characterization of the cistrome. Previously, DNA microarray was also used (ChIP-on-chip or ChIP-chip).
RNP immunoprecipitation (RIP)
Similar to chromatin immunoprecipitation (ChIP) outlined above, but rather than targeting DNA binding proteins as in ChIP, an RNP immunoprecipitation targets ribonucleoproteins (RNPs).[2] Live cells are first lysed and then the target protein and associated RNA are immunoprecipitated using an antibody targeting the protein of interest. The purified RNA-protein complexes can be separated by performing an RNA extraction and the identity of the RNA can be determined by cDNA sequencing[3] or RT-PCR. Some variants of RIP, such as PAR-CLIP include cross-linking steps, which then require less careful lysis conditions.
Tagged proteins
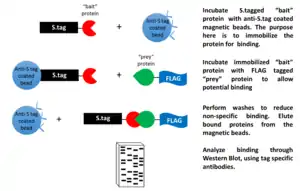
One of the major technical hurdles with immunoprecipitation is the great difficulty in generating an antibody that specifically targets a single known protein. To get around this obstacle, many groups will engineer tags onto either the C- or N- terminal end of the protein of interest. The advantage here is that the same tag can be used time and again on many different proteins and the researcher can use the same antibody each time. The advantages with using tagged proteins are so great that this technique has become commonplace for all types of immunoprecipitation including all of the types of IP detailed above. Examples of tags in use are the green fluorescent protein (GFP) tag, glutathione-S-transferase (GST) tag and the FLAG-tag tag. While the use of a tag to enable pull-downs is convenient, it raises some concerns regarding biological relevance because the tag itself may either obscure native interactions or introduce new and unnatural interactions.
Methods
The two general methods for immunoprecipitation are the direct capture method and the indirect capture method.
Direct
Antibodies that are specific for a particular protein (or group of proteins) are immobilized on a solid-phase substrate such as superparamagnetic microbeads or on microscopic agarose (non-magnetic) beads. The beads with bound antibodies are then added to the protein mixture, and the proteins that are targeted by the antibodies are captured onto the beads via the antibodies; in other words, they become immunoprecipitated.
Indirect
Antibodies that are specific for a particular protein, or a group of proteins, are added directly to the mixture of protein. The antibodies have not been attached to a solid-phase support yet. The antibodies are free to float around the protein mixture and bind their targets. As time passes, beads coated in Protein A/G are added to the mixture of antibody and protein. At this point, the antibodies, which are now bound to their targets, will stick to the beads.
From this point on, the direct and indirect protocols converge because the samples now have the same ingredients. Both methods give the same end-result with the protein or protein complexes bound to the antibodies which themselves are immobilized onto the beads.
Selection
An indirect approach is sometimes preferred when the concentration of the protein target is low or when the specific affinity of the antibody for the protein is weak. The indirect method is also used when the binding kinetics of the antibody to the protein is slow for a variety of reasons. In most situations, the direct method is the default, and the preferred, choice.
Technological advances
Agarose
Historically the solid-phase support for immunoprecipitation used by the majority of scientists has been highly-porous agarose beads (also known as agarose resins or slurries). The advantage of this technology is a very high potential binding capacity, as virtually the entire sponge-like structure of the agarose particle (50 to 150μm in size) is available for binding antibodies (which will in turn bind the target proteins) and the use of standard laboratory equipment for all aspects of the IP protocol without the need for any specialized equipment. The advantage of an extremely high binding capacity must be carefully balanced with the quantity of antibody that the researcher is prepared to use to coat the agarose beads. Because antibodies can be a cost-limiting factor, it is best to calculate backward from the amount of protein that needs to be captured (depending upon the analysis to be performed downstream), to the amount of antibody that is required to bind that quantity of protein (with a small excess added in order to account for inefficiencies of the system), and back still further to the quantity of agarose that is needed to bind that particular quantity of antibody. In cases where antibody saturation is not required, this technology is unmatched in its ability to capture extremely large quantities of captured target proteins. The caveat here is that the "high capacity advantage" can become a "high capacity disadvantage" that is manifested when the enormous binding capacity of the sepharose/agarose beads is not completely saturated with antibodies. It often happens that the amount of antibody available to the researcher for their immunoprecipitation experiment is less than sufficient to saturate the agarose beads to be used in the immunoprecipitation. In these cases the researcher can end up with agarose particles that are only partially coated with antibodies, and the portion of the binding capacity of the agarose beads that is not coated with antibody is then free to bind anything that will stick, resulting in an elevated background signal due to non-specific binding of lysate components to the beads, which can make data interpretation difficult. While some may argue that for these reasons it is prudent to match the quantity of agarose (in terms of binding capacity) to the quantity of antibody that one wishes to be bound for the immunoprecipitation, a simple way to reduce the issue of non-specific binding to agarose beads and increase specificity is to preclear the lysate, which for any immunoprecipitation is highly recommended.[4][5]
Preclearing
Lysates are complex mixtures of proteins, lipids, carbohydrates and nucleic acids, and one must assume that some amount of non-specific binding to the IP antibody, Protein A/G or the beaded support will occur and negatively affect the detection of the immunoprecipitated target(s). In most cases, preclearing the lysate at the start of each immunoprecipitation experiment (see step 2 in the "protocol" section below)[6] is a way to remove potentially reactive components from the cell lysate prior to the immunoprecipitation to prevent the non-specific binding of these components to the IP beads or antibody. The basic preclearing procedure is described below, wherein the lysate is incubated with beads alone, which are then removed and discarded prior to the immunoprecipitation.[6] This approach, though, does not account for non-specific binding to the IP antibody, which can be considerable. Therefore, an alternative method of preclearing is to incubate the protein mixture with exactly the same components that will be used in the immunoprecipitation, except that a non-target, irrelevant antibody of the same antibody subclass as the IP antibody is used instead of the IP antibody itself.[5] This approach attempts to use as close to the exact IP conditions and components as the actual immunoprecipitation to remove any non-specific cell constituent without capturing the target protein (unless, of course, the target protein non-specifically binds to some other IP component, which should be properly controlled for by analyzing the discarded beads used to preclear the lysate). The target protein can then be immunoprecipitated with the reduced risk of non-specific binding interfering with data interpretation.
Superparamagnetic beads
While the vast majority of immunoprecipitations are performed with agarose beads, the use of superparamagnetic beads for immunoprecipitation is a newer approach that is gaining in popularity as an alternative to agarose beads for IP applications. Unlike agarose, magnetic beads are solid and can be spherical, depending on the type of bead, and antibody binding is limited to the surface of each bead. While these beads do not have the advantage of a porous center to increase the binding capacity, magnetic beads are significantly smaller than agarose beads (1 to 4μm), and the greater number of magnetic beads per volume than agarose beads collectively gives magnetic beads an effective surface area-to-volume ratio for optimum antibody binding.
Commercially available magnetic beads can be separated based by size uniformity into monodisperse and polydisperse beads. Monodisperse beads, also called microbeads, exhibit exact uniformity, and therefore all beads exhibit identical physical characteristics, including the binding capacity and the level of attraction to magnets. Polydisperse beads, while similar in size to monodisperse beads, show a wide range in size variability (1 to 4μm) that can influence their binding capacity and magnetic capture. Although both types of beads are commercially available for immunoprecipitation applications, the higher quality monodisperse superparamagnetic beads are more ideal for automatic protocols because of their consistent size, shape and performance. Monodisperse and polydisperse superparamagnetic beads are offered by many companies, including Invitrogen, Thermo Scientific, and Millipore.
Agarose vs. magnetic beads
Proponents of magnetic beads claim that the beads exhibit a faster rate of protein binding[7][8][9] over agarose beads for immunoprecipitation applications, although standard agarose bead-based immunoprecipitations have been performed in 1 hour.[5] Claims have also been made that magnetic beads are better for immunoprecipitating extremely large protein complexes because of the complete lack of an upper size limit for such complexes,[7][8][10] although there is no unbiased evidence stating this claim. The nature of magnetic bead technology does result in less sample handling[8] due to the reduced physical stress on samples of magnetic separation versus repeated centrifugation when using agarose, which may contribute greatly to increasing the yield of labile (fragile) protein complexes.[8][9][10] Additional factors, though, such as the binding capacity, cost of the reagent, the requirement of extra equipment and the capability to automate IP processes should be considered in the selection of an immunoprecipitation support.
Binding capacity
Proponents of both agarose and magnetic beads can argue whether the vast difference in the binding capacities of the two beads favors one particular type of bead. In a bead-to-bead comparison, agarose beads have significantly greater surface area and therefore a greater binding capacity than magnetic beads due to the large bead size and sponge-like structure. But the variable pore size of the agarose causes a potential upper size limit that may affect the binding of extremely large proteins or protein complexes to internal binding sites, and therefore magnetic beads may be better suited for immunoprecipitating large proteins or protein complexes than agarose beads, although there is a lack of independent comparative evidence that proves either case.
Some argue that the significantly greater binding capacity of agarose beads may be a disadvantage because of the larger capacity of non-specific binding. Others may argue for the use of magnetic beads because of the greater quantity of antibody required to saturate the total binding capacity of agarose beads, which would obviously be an economical disadvantage of using agarose. While these arguments are correct outside the context of their practical use, these lines of reasoning ignore two key aspects of the principle of immunoprecipitation that demonstrates that the decision to use agarose or magnetic beads is not simply determined by binding capacity.
First, non-specific binding is not limited to the antibody-binding sites on the immobilized support; any surface of the antibody or component of the immunoprecipitation reaction can bind to nonspecific lysate constituents, and therefore nonspecific binding will still occur even when completely saturated beads are used. This is why it is important to preclear the sample before the immunoprecipitation is performed.
Second, the ability to capture the target protein is directly dependent upon the amount of immobilized antibody used, and therefore, in a side-by-side comparison of agarose and magnetic bead immunoprecipitation, the most protein that either support can capture is limited by the amount of antibody added. So the decision to saturate any type of support depends on the amount of protein required, as described above in the Agarose section of this page.
Cost
The price of using either type of support is a key determining factor in using agarose or magnetic beads for immunoprecipitation applications. A typical first-glance calculation on the cost of magnetic beads compared to sepharose beads may make the sepharose beads appear less expensive. But magnetic beads may be competitively priced compared to agarose for analytical-scale immunoprecipitations depending on the IP method used and the volume of beads required per IP reaction.
Using the traditional batch method of immunoprecipitation as listed below, where all components are added to a tube during the IP reaction, the physical handling characteristics of agarose beads necessitate a minimum quantity of beads for each IP experiment (typically in the range of 25 to 50μl beads per IP). This is because sepharose beads must be concentrated at the bottom of the tube by centrifugation and the supernatant removed after each incubation, wash, etc. This imposes absolute physical limitations on the process, as pellets of agarose beads less than 25 to 50μl are difficult if not impossible to visually identify at the bottom of the tube. With magnetic beads, there is no minimum quantity of beads required due to magnetic handling, and therefore, depending on the target antigen and IP antibody, it is possible to use considerably less magnetic beads.
Conversely, spin columns may be employed instead of normal microfuge tubes to significantly reduce the amount of agarose beads required per reaction. Spin columns contain a filter that allows all IP components except the beads to flow through using a brief centrifugation and therefore provide a method to use significantly less agarose beads with minimal loss.
Equipment
As mentioned above, only standard laboratory equipment is required for the use of agarose beads in immunoprecipitation applications, while high-power magnets are required for magnetic bead-based IP reactions. While the magnetic capture equipment may be cost-prohibitive, the rapid completion of immunoprecipitations using magnetic beads may be a financially beneficial approach when grants are due, because a 30-minute protocol with magnetic beads compared to overnight incubation at 4 °C with agarose beads may result in more data generated in a shorter length of time.[7][8][9]
Automation
An added benefit of using magnetic beads is that automated immunoprecipitation devices are becoming more readily available. These devices not only reduce the amount of work and time to perform an IP, but they can also be used for high-throughput applications.
Summary
While clear benefits of using magnetic beads include the increased reaction speed, more gentle sample handling and the potential for automation, the choice of using agarose or magnetic beads based on the binding capacity of the support medium and the cost of the product may depend on the protein of interest and the IP method used. As with all assays, empirical testing is required to determine which method is optimal for a given application.
Protocol
Background
Once the solid substrate bead technology has been chosen, antibodies are coupled to the beads and the antibody-coated-beads can be added to the heterogeneous protein sample (e.g. homogenized tissue). At this point, antibodies that are immobilized to the beads will bind to the proteins that they specifically recognize. Once this has occurred the immunoprecipitation portion of the protocol is actually complete, as the specific proteins of interest are bound to the antibodies that are themselves immobilized to the beads. Separation of the immunocomplexes from the lysate is an extremely important series of steps, because the protein(s) must remain bound to each other (in the case of co-IP) and bound to the antibody during the wash steps to remove non-bound proteins and reduce background.
When working with agarose beads, the beads must be pelleted out of the sample by briefly spinning in a centrifuge with forces between 600–3,000 x g (times the standard gravitational force). This step may be performed in a standard microcentrifuge tube, but for faster separation, greater consistency and higher recoveries, the process is often performed in small spin columns with a pore size that allows liquid, but not agarose beads, to pass through. After centrifugation, the agarose beads will form a very loose fluffy pellet at the bottom of the tube. The supernatant containing contaminants can be carefully removed so as not to disturb the beads. The wash buffer can then be added to the beads and after mixing, the beads are again separated by centrifugation.
With superparamagnetic beads, the sample is placed in a magnetic field so that the beads can collect on the side of the tube. This procedure is generally complete in approximately 30 seconds, and the remaining (unwanted) liquid is pipetted away. Washes are accomplished by resuspending the beads (off the magnet) with the washing solution and then concentrating the beads back on the tube wall (by placing the tube back on the magnet). The washing is generally repeated several times to ensure adequate removal of contaminants. If the superparamagnetic beads are homogeneous in size and the magnet has been designed properly, the beads will concentrate uniformly on the side of the tube and the washing solution can be easily and completely removed.
After washing, the precipitated protein(s) are eluted and analyzed by gel electrophoresis, mass spectrometry, western blotting, or any number of other methods for identifying constituents in the complex. Protocol times for immunoprecipitation vary greatly due to a variety of factors, with protocol times increasing with the number of washes necessary or with the slower reaction kinetics of porous agarose beads.
Steps
- Lyse cells and prepare sample for immunoprecipitation.
- Pre-clear the sample by passing the sample over beads alone or bound to an irrelevant antibody to soak up any proteins that non-specifically bind to the IP components.
- Incubate solution with antibody against the protein of interest. Antibody can be attached to solid support before this step (direct method) or after this step (indirect method). Continue the incubation to allow antibody-antigen complexes to form.
- Precipitate the complex of interest, removing it from bulk solution.
- Wash precipitated complex several times. Spin each time between washes when using agarose beads or place tube on magnet when using superparamagnetic beads and then remove the supernatant. After the final wash, remove as much supernatant as possible.
- Elute proteins from the solid support using low-pH or SDS sample loading buffer.
- Analyze complexes or antigens of interest. This can be done in a variety of ways:
- SDS-PAGE (sodium dodecyl sulfate-polyacrylamide gel electrophoresis) followed by gel staining.
- SDS-PAGE followed by: gel staining, cutting out individual stained protein bands, and sequencing the proteins in the bands by matrix-assisted laser desorption/ionization (MALDI) mass spectrometry.
- Transfer and Western Blot using another antibody for proteins that were interacting with the antigen, followed by detection using a chemiluminescent or fluorescent secondary antibody.
References
- Rashid, K. A., Hevi, S., Chen, Y., Le Cahérec, F., & Chuck, S. L. (2002). A Proteomic Approach Identifies Proteins in Hepatocytes That Bind Nascent Apolipoprotein B. The Journal of Biological Chemistry, 277(24), 22010–22017. https://doi.org/10.1074/jbc.M112448200
- Keene JD, Komisarow JM, Friedersdorf MB (2006). "RIP-Chip: the isolation and identification of mRNAs, microRNAs and protein components of ribonucleoprotein complexes from cell extracts". Nat Protoc. 1 (1): 302–7. doi:10.1038/nprot.2006.47. PMID 17406249. S2CID 25925403.
- Sanford JR, Wang X, Mort M, et al. (March 2009). "Splicing factor SFRS1 recognizes a functionally diverse landscape of RNA transcripts". Genome Res. 19 (3): 381–94. doi:10.1101/gr.082503.108. PMC 2661799. PMID 19116412.
- Bonifacino, J. S., Dell'Angelica, E. C. and Springer, T. A. 2001. Immunoprecipitation. Current Protocols in Molecular Biology. 10.16.1–10.16.29.
- Rosenberg, Ian (2005). Protein analysis and purification: benchtop techniques. Springer. p. 520. ISBN 978-0-8176-4340-9.
- Crowell RE, Du Clos TW, Montoya G, Heaphy E, Mold C (November 1991). "C-reactive protein receptors on the human monocytic cell line U-937. Evidence for additional binding to Fc gamma RI". Journal of Immunology. 147 (10): 3445–51. PMID 1834740.
- Alber F, Dokudovskaya S, Veenhoff LM, et al. (November 2007). "The molecular architecture of the nuclear pore complex". Nature. 450 (7170): 695–701. Bibcode:2007Natur.450..695A. doi:10.1038/nature06405. PMID 18046406. S2CID 4431057.
- Alber F, Dokudovskaya S, Veenhoff LM, et al. (November 2007). "Determining the architectures of macromolecular assemblies". Nature. 450 (7170): 683–94. Bibcode:2007Natur.450..683A. doi:10.1038/nature06404. PMID 18046405. S2CID 2171750.
- Cristea IM, Williams R, Chait BT, Rout MP (December 2005). "Fluorescent proteins as proteomic probes". Molecular & Cellular Proteomics. 4 (12): 1933–41. doi:10.1074/mcp.M500227-MCP200. PMID 16155292.
- Niepel M, Strambio-de-Castillia C, Fasolo J, Chait BT, Rout MP (July 2005). "The nuclear pore complex–associated protein, Mlp2p, binds to the yeast spindle pole body and promotes its efficient assembly". The Journal of Cell Biology. 170 (2): 225–35. doi:10.1083/jcb.200504140. PMC 2171418. PMID 16027220.
External links
| Library resources about Immunoprecipitation |
- Analysis of Proteins Using Immunoprecipitation at ufl.edu
- Immunoprecipitation at the US National Library of Medicine Medical Subject Headings (MeSH)
- Chromatin+immunoprecipitation at the US National Library of Medicine Medical Subject Headings (MeSH)
- Introduction to Immunoprecipitation Methodology